
Article | 04/12/2024
Comment se forment les cristaux ? Du bécher à la croissance hydrothermale
04/12/2024
Auteur(s) / Autrice(s) :
Publié par :
- Olivier DequinceyENS de Lyon / DGESCO
Résumé
Principes et modalités de la cristallisation à partir d’une expérience simple. Application aux cristallisations de surface et dans les filons hydrothermaux.

Source - © 2023 — D'après Thomas Schüpbach, Ipsach / Kristalle
L’anatase est un dioxyde de titane TiO2. Les plus longs cristaux de quartz ont une taille de l’ordre du centimètre.
La majorité des cristaux bien constitués que l’on voit dans la nature se sont formés à partir d’une solution liquide. Pour comprendre les principes qui gouvernent la formation de ces cristaux, nous prendrons, dans un premier temps, l'exemple de la création d'un cristal de sulfate de cuivre, expérience réalisable chez soi. Puis nous appliquerons les concepts rencontrés à d'autres minéraux facilement solubles que l'on trouve dans la nature : gypse et calcite. Ensuite nous aborderont les “superpouvoirs” de l'eau et comment des minéraux comme le quartz peuvent se former. Enfin, nous explorerons la formation des filons et des géodes où se fabriquent les plus beaux cristaux.
Faire cristalliser du sulfate de cuivre pour s'initier aux bases de la cristallisation
Pour fixer les idées appelons “Blue” ce cristal bleu de sulfate de cuivre pentahydraté (CuSO4·5H2O). De multiples recettes peuvent être trouvées sur le web pour le fabriquer (Croissance de minéral – Cultivez les cristaux | The Best Way to Grow Big Copper Sulfate Crystals | How to grow a large crystal of copper (II) sulphate in 5 days | Copper Sulfate Crystals Recipe), nous rappelons ici le matériel et le déroulement de la manipulation.
Ingrédients et matériel nécessaires
Eau distillée / déminéralisée de préférence (afin d'éviter la présence d'autres ions susceptibles de cristalliser).
Sulfate de cuivre pentahydraté : CuSO4·5H2O en poudre (en droguerie, magasin de bricolage, jardinerie…) [produit irritant et nocif si on l'avale ; utiliser lunettes, blouse et gants pour manipuler].
Casserole pour faire chauffer l'eau.
Pot en verre, style bécher, qui résiste à l'eau bouillante.
Coupelle en verre ou vieux bol.
Crayon (ou tige de métal ou de verre), fil fin, par exemple de nylon.
Papier filtre.
Pince à épiler (pour enlever les germes “excédentaires”).
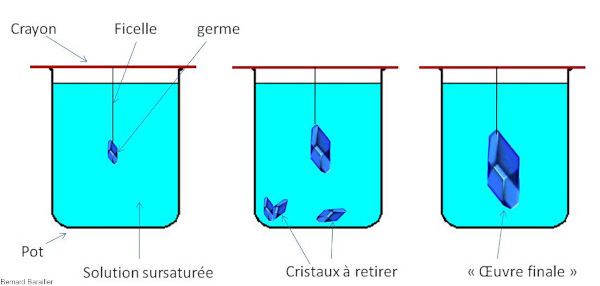
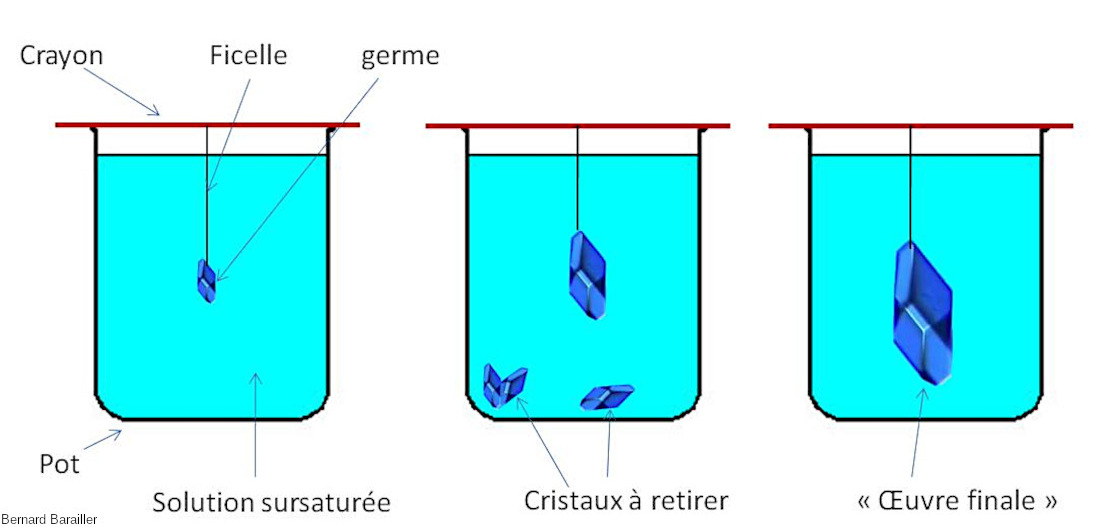
Source - © 2024 — Bernard Barailler
Recette
- Faire chauffer de l'eau jusqu'à l'ébullition (100°C).
- Verser cette eau “bouillante” dans le pot en verre. Ajouter le sulfate de cuivre en remuant. Continuer tant que vous pouvez le dissoudre. Quand vous n'y arrivez plus et qu'il reste du sulfate de cuivre solide dans votre bain, la solution est dite sursaturée.
- Verser alors une partie du liquide (sans la partie solide) dans une coupelle et laisser reposer dans un endroit calme. Réserver le reste de la solution pour plus tard.
- Dès que l'eau dans la coupelle s'est évaporée, sélectionner parmi les cristaux qui s'y sont déposés le plus beau. Ce sera votre germe.
- Fixer le germe sur une ficelle, et la ficelle à un crayon / une tige.
- À partir du reste de la solution initiale, refaire une solution sursaturée c’est-à-dire dans laquelle vous ne pouvez plus dissoudre de sulfate de cuivre.
- Filtrer la solution pour enlever toute impureté.
- Verser la nouvelle solution dans un pot si possible entourée de sac isotherme (pour que la température s'abaisse lentement), sur une table stable (sans vibration), avec un papier filtre dessus (pour éviter que tombent des poussières susceptibles de créer des germes concurrents), à l'abri des courants d'air (pour éviter la convection forcée).
- Tremper le germe dans la solution (figure 2).
- Laisser reposer et regarder régulièrement le pot.
- Si d'autres cristaux apparaissent, verser la solution sans ces cristaux dans un autre pot et remettre le cristal au bout de la ficelle (afin d'éviter la concurrence des autres germes lors de la croissance cristalline).
- Surveiller et répéter l'opération jusqu'à l'obtention d'un beau cristal bleu (durée de la journée à plusieurs jours voire semaines selon la taille désirée et la quantité initiale). Le retirer et le laisser sécher.
Pour conserver votre cristal le plus longtemps possible, mettez-le sous cloche pendu à son fil et scellez les bords avec de la bougie (pour éviter tout contact avec l'humidité ambiante susceptible d'abimer votre cristal qui est soluble).

Source - © 2024 — D'après Cristalverse / Xhttps://x.com/thecrystalverse
Résultat au bout de 2 mois de manipulation. Film sur YouTube, chaine Crystalverse(lien externe - nouvelle fenêtre). Autre exemple filmé sur 2 jours par le CiMF(lien externe - nouvelle fenêtre).
Quand on essaye de faire des cristaux comme Blue, plusieurs constatations s'imposent.
- Concernant la dissolution, on s'aperçoit que si on met beaucoup de sulfate de cuivre dans de l'eau à une température fixée, au bout d'un moment le sulfate de cuivre qu'on ajoute n'arrive plus à se dissoudre. Mais si on rajoute de l'eau, de nouveau le sulfate de cuivre se dissout. Donc la concentration de sulfate de cuivre dans l'eau est un facteur de dissolution important à prendre en compte.
- Par ailleurs, on peut s'apercevoir que pour la même quantité d'eau, plus on la chauffe plus on peut y mettre du sulfate de cuivre dissout. Donc la température est un deuxième facteur qui permet d'augmenter la dissolution du sulfate de cuivre.
- À l'inverse, quand on laisse s'évaporer une solution contenant des ions sulfate et des ions cuivre, des cristaux bleus apparaissent au bout d'un moment. Si la température baisse, des cristaux peuvent aussi apparaitre. On a donc bien une relation réversible entre la cristallisation et la dissolution.
- En termes de cinétique (vitesse de la réaction), on peut s'apercevoir que si on refroidit rapidement on aura beaucoup de petits cristaux. À contrario, si on refroidit très lentement, on pourra voir croitre des cristaux plus grands.
- Finalement, on peut s'apercevoir expérimentalement que si on met un germe d'un cristal d'une taille importante, il va croitre fortement au détriment des autres plus petits.
Approche microscopique simplifiée de la cristallisation
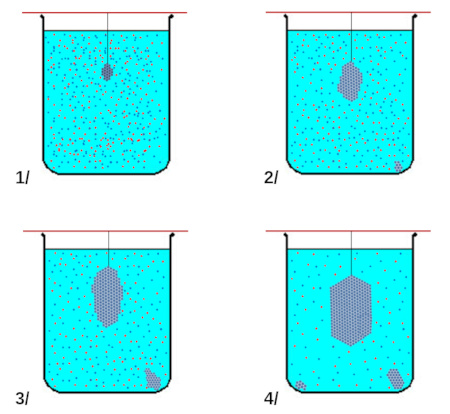
Source - © 2024 — Bernard Barailler
Cristallisation de sulfate de cuivre
Quand on fait l'expérience de la croissance du sulfate de cuivre, on s'aperçoit qu'il y a plusieurs étapes dans la croissance des cristaux à partir d'un fluide. On a d'abord la production des germes, puis la concurrence entre ces germes pour récupérer le maximum d'ions lors de leur croissance cristalline. Les gros cristaux croissent plus vite que les petits en captant les ions présents en solution.
Dans le cas de Blue, les ions “bleus” Cu2+ et “rouges” SO42− ainsi que les molécules d'eau, H2O, vont s'assembler pour former une maille élémentaire (rond violet) des cristaux de sulfate de cuivre pentahydraté (CuSO4·5H2O). Ce sont les unités de croissance. La solution s'appauvrit en ions au fur et à mesure que les cristaux croissent, jusqu'à ce que la sursaturation cesse (figure ci-dessus).
À contrario, si on trempe un cristal dans une solution non sursaturée, c'est-à-dire une solution dans laquelle on peut encore dissoudre des cristaux, alors le germe se dissout jusqu'à ce que la solution soit au bord de la sursaturation.
Si vous préférez manipuler du sucre…
Pour limiter les précautions à prendre avec des élèves ou étudiants (il reste la manipulation d’eau chaude voire bouillante), on peut expérimenter la cristallisation en reprenant le protocole décrit pour le sulfate de cuivre mais en utilisant du sucre. On “fabrique” alors des cristaux de sucre candi, voir, par exemple, Cristallisation du sucre ou 3 manières de faire du sucre candi. Des variations temporaires avec des colorants alimentaires permettent même de visualiser des “couches de croissance”.
Du soluté au cristal : une réaction chimique réversible
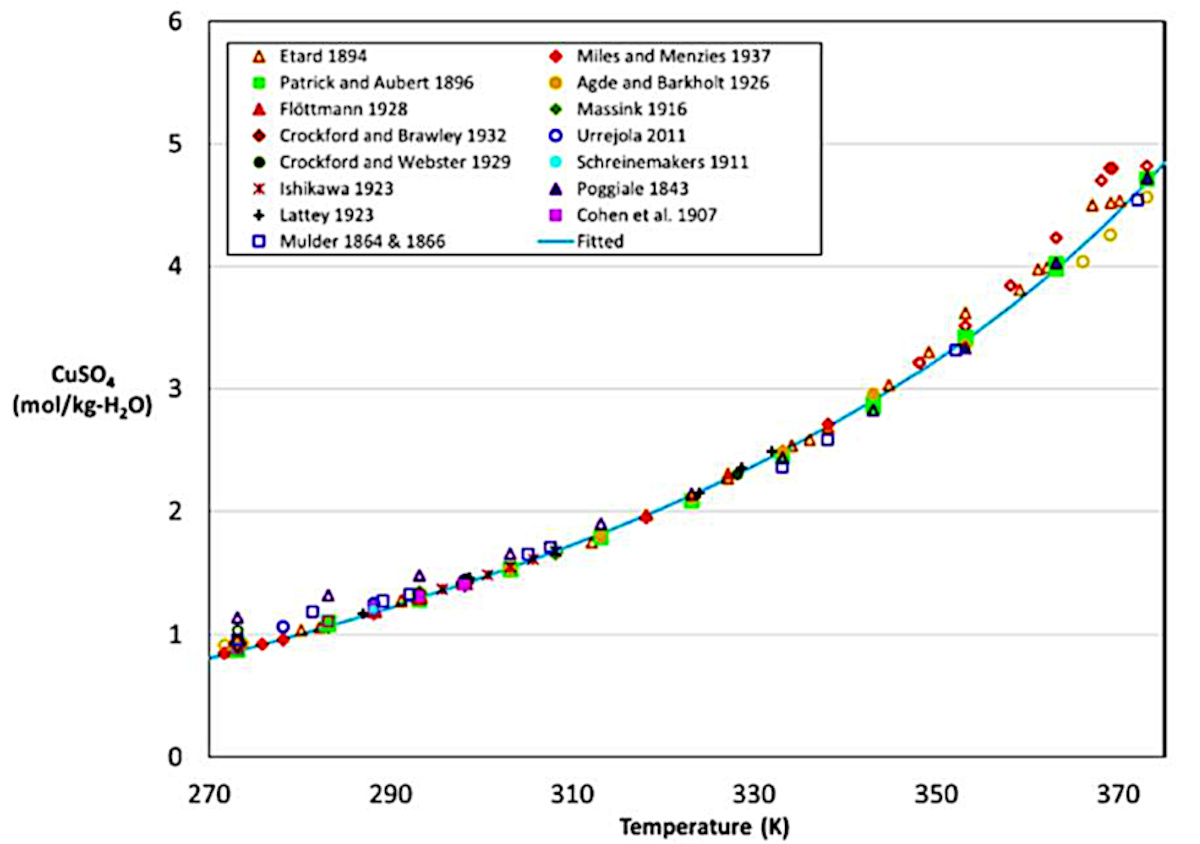
Depuis longtemps les scientifiques ont mesuré les conditions de dissolution du sulfate de cuivre.
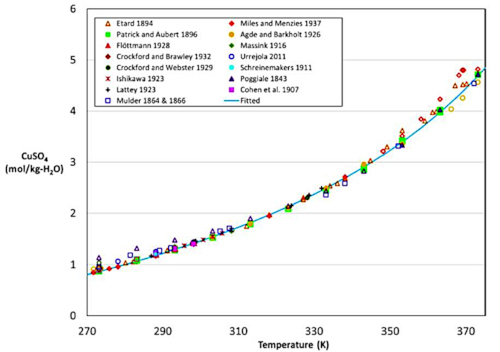
Source - © 2022 — D’après Sibarini et al. [ 25 ]
Ils ont contribué à tracer la courbe de solubilité du sulfate de cuivre. La courbe de solubilité (ou courbe de saturation) donne la limite de solubilité d'un sel en fonction de la température. C'est une courbe d'équilibre[1].
La courbe de solubilité est obtenue expérimentalement, pour un composé donné, en partant d'une solution non saturée et en rajoutant progressivement du sel. La limite de solubilité est obtenue lorsqu'il n'y a plus dissolution. Il s'agit de voir pour chaque température T quel est la concentration limite C du sulfate de cuivre dans l'eau. On trace alors la courbe C(T) (figure 4). Pour être précis, il s'agit de la solubilité dans de l'eau pure à pH neutre sous une pression de 1 atm.
À droite de cette courbe (figure 5), le soluté composé des ions en solution est dans une phase liquide. Les ions peuvent se mouvoir librement au sein du solvant (ici l'eau). À gauche de la courbe (figure 5), les atomes sont figés dans la structure solide du cristal. Leurs positions sont fixes dans celui-ci.
Quand on franchit la courbe de solubilité, il s'opère une transition de phase :
de droite à gauche on passe de l'état liquide à l'état solide, il y a cristallisation ;
de gauche à droite on passe de l'état solide à l'état liquide, il y a dissolution.
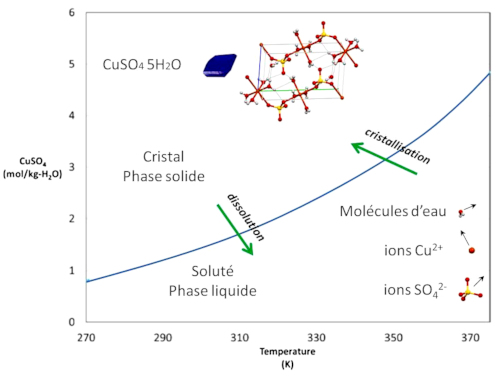
Source - © 2024 — Bernard Barailler – Ben Mills (2022) / wikimedia (modèle moléculaire)
Cu en orange, S en jaune, O en rouge et H en gris.
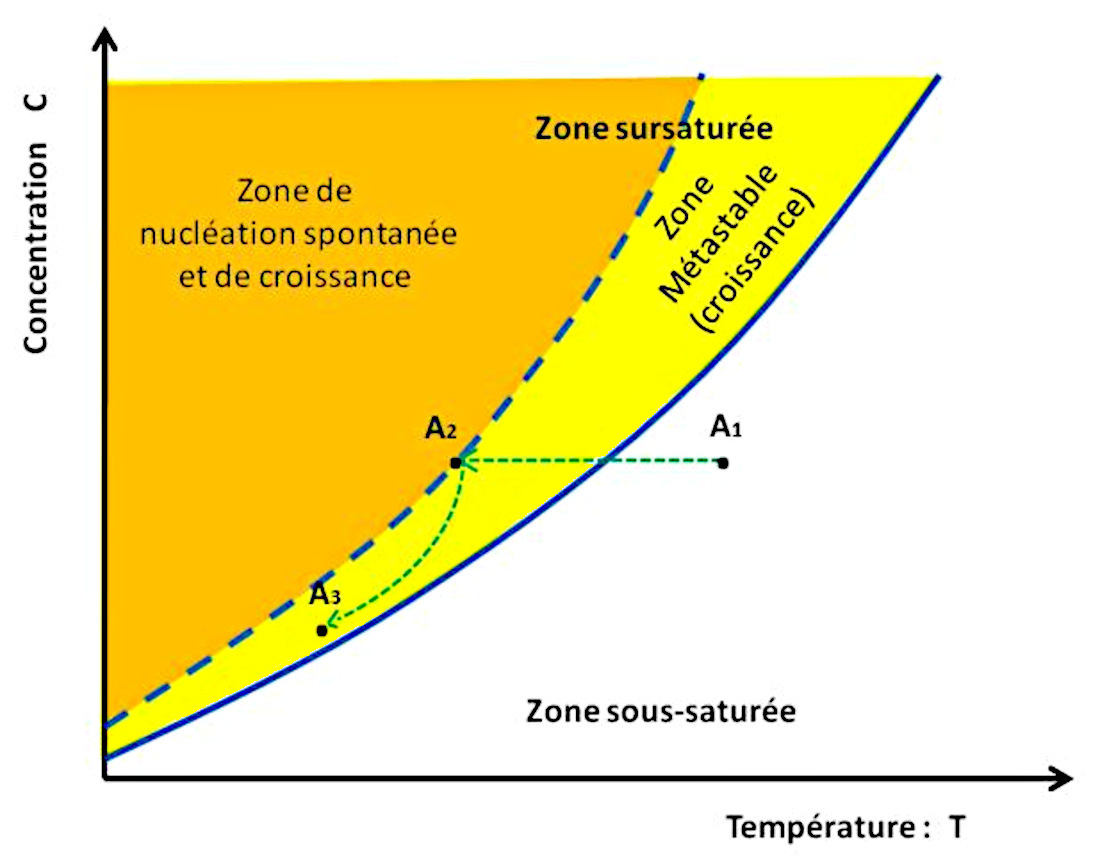
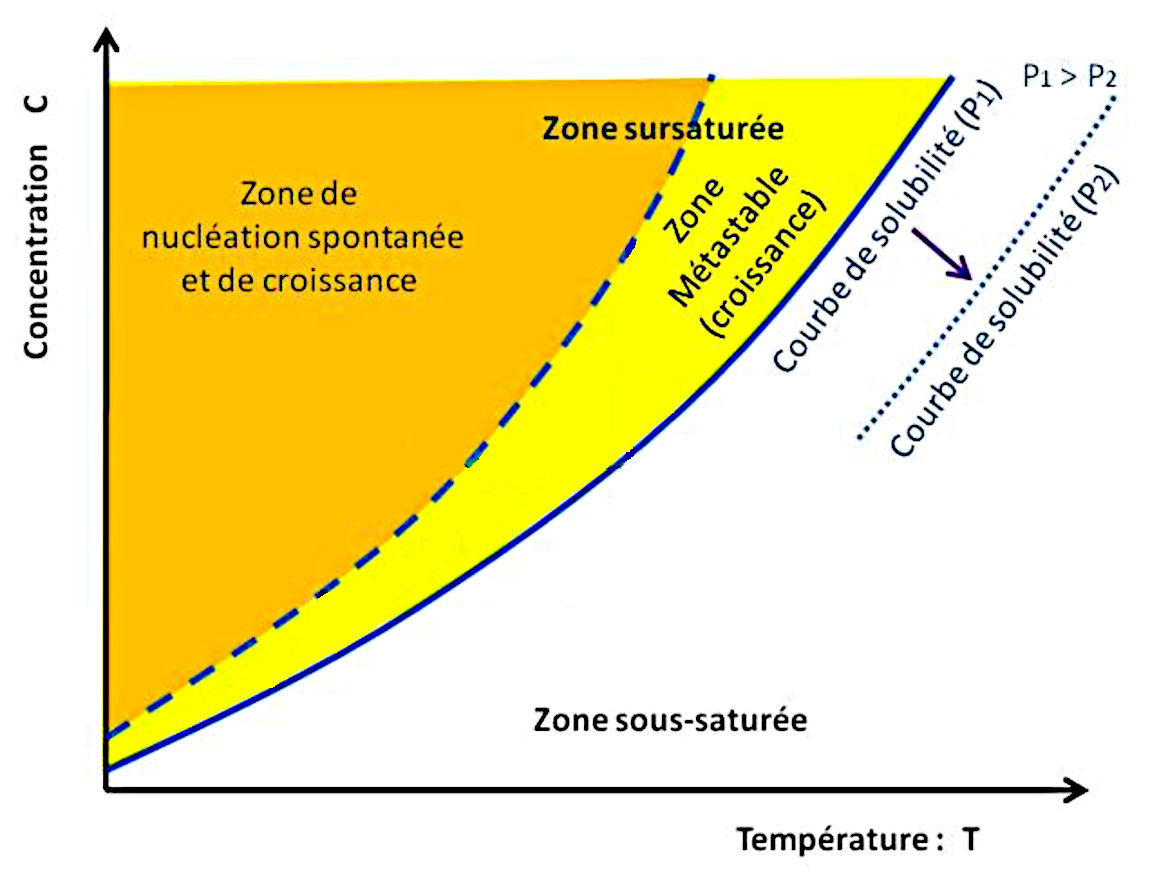
De façon générale, il faut tenir compte d’une autre courbe importante, en pointillés bleus sur les figures 6, 7 et 10 : la courbe de nucléation spontanée (ou courbe de sursaturation). La nucléation correspondant à l'étape de création des premiers germes, des premiers cristaux solides. Cette courbe de nucléation spontanée marque la limite au-delà de laquelle la solution donne naissance spontanément à des germes de cristaux, initiant ainsi la cristallisation.
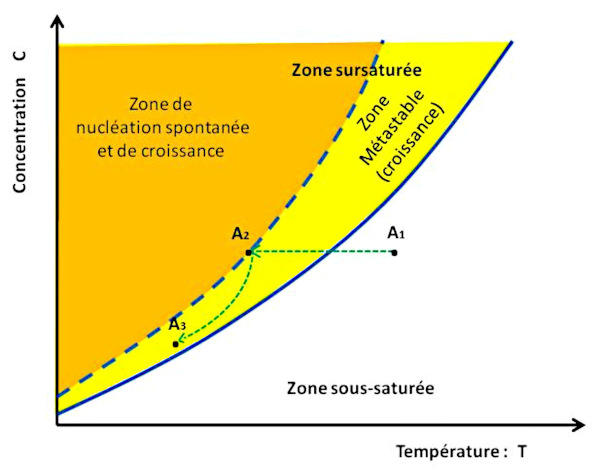
Source - © 2024 — Bernard Barailler
On suit le trajet d'une solution A1 “chaude” qui se refroidit jusqu'à A3. Dans un premier temps, de A1 à A2, la température baisse à concentration constante donc en l'absence de formation de cristaux. En effet, en l'absence de germes initiaux, la cristallisation ne débute pas au passage de la courbe de solubilité (saturation) mais seulement lorsqu'elle atteint, en A2, la courbe de nucléation spontanée (sursaturation). Ensuite, la croissance de cristaux appauvrit la solution dont la température baisse encore, et la croissance cristalline continue tant que la solution reste dans la zone métastable, entre courbes de saturation et de sursaturation.
Ces deux courbes délimitent trois zones distinctes :
La zone sous-saturée, à droite de la courbe de solubilité : dans cette zone, la solution n'est pas saturée, et elle peut encore dissoudre du sel.
La zone métastable, entre la courbe de solubilité et la courbe de nucléation spontanée : en présence de germes, il y a cristallisation par grossissement et apport d'unités de croissance. En l'absence totale de germes, la solution peut rester avec une seule phase liquide, sans nucléation et donc sans cristallisation. Cette notion d'état métastable est la même que l'eau liquide dont on abaisse la température en dessous de 0°C sans vibration, ni germe ni poussière. L'eau reste alors dans l'état liquide, mais la moindre perturbation la transforme brutalement en glace. Dans le cas de la figure 6, on baisse la température sans évaporation (concentration constante). La solution évolue du point A1 au point A2 sans cristallisation.
La zone dite labile, à gauche de la courbe de nucléation spontanée : la nucléation a lieu spontanément, les germes ainsi formés de développent par adjonction d'unités de croissance, diminuant ainsi la concentration du liquide. La solution va alors rejoindre la courbe de solubilité à une vitesse dépendant des conditions opératoires (flux des unités de croissance, évacuation de la chaleur de la réaction, cinétique de grossissement et de nucléation…). Dans la figure 6, la solution, dont des ions sont soustraits du fait de la cristallisation, suit le trajet de A2 à A3.
Dans la figure 6, le trajet suivi (en vert) correspond à une baisse de la température, initialement à concentration constante (c'est-à-dire, par exemple, sans évaporation).
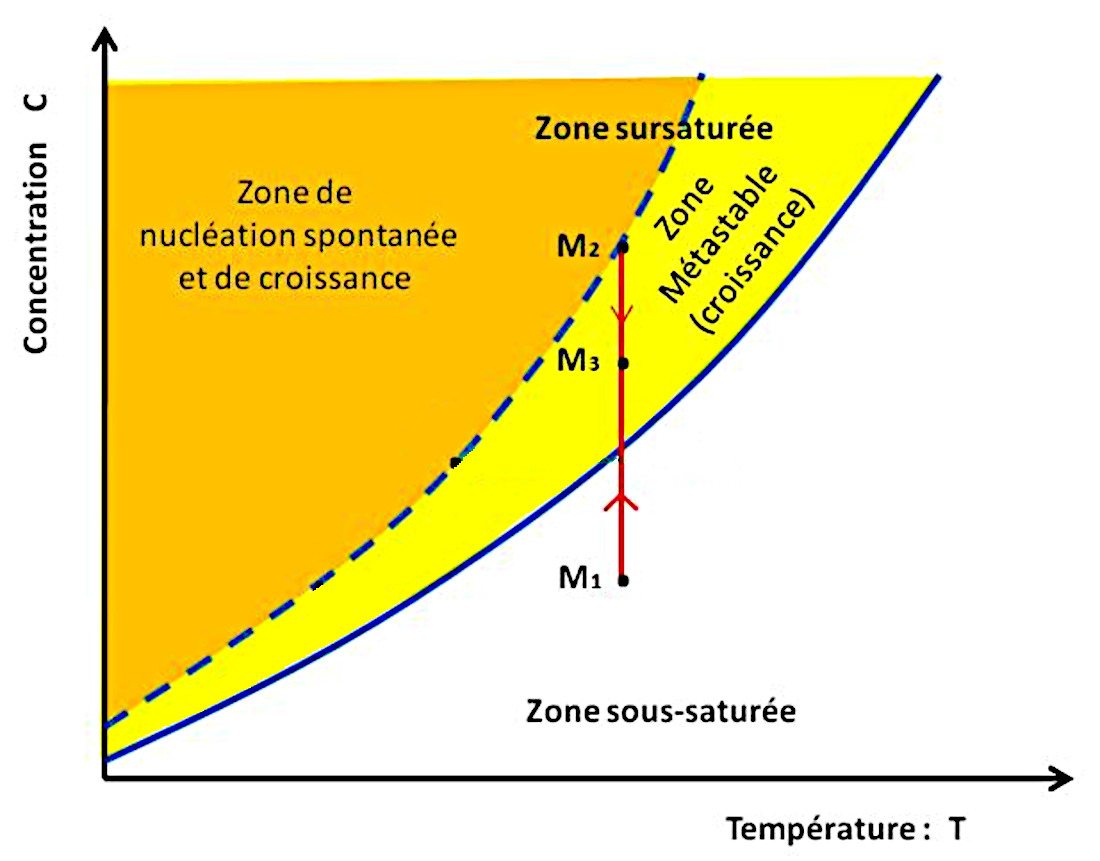
La figure 7 montre un trajet (en rouge) à température constante (dans un calorimètre par exemple) avec une évaporation progressive. La concentration augmente progressivement jusqu'à ce qu'un cristal se développe lorsque la courbe de sursaturation est atteinte, et capte des unités de croissance. La concentration du soluté restant baisse alors. On passe de M1 à M2 puis on redescend, par exemple vers M3.
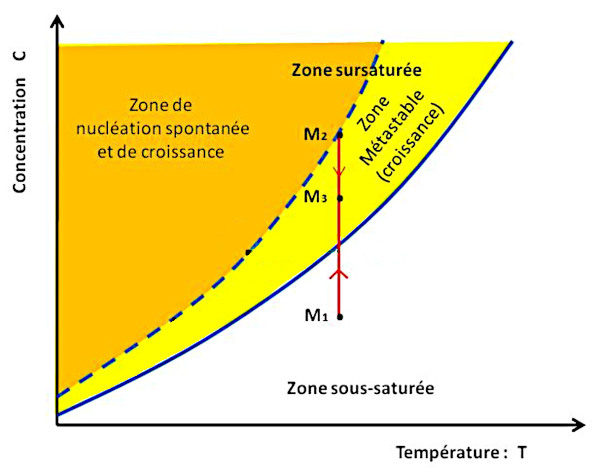
Source - © 2024 — Bernard Barailler
On suit le trajet d'une solution M1 maintenue à température constante mais soumise à évaporation. De M1 à M2, la concentration augmente jusqu'à ce que, en l'absence de germes initiaux, la solution arrive à sursaturation et que des cristaux se développent. Alors, de M2 à M3 la croissance cristalline “pompe” des ions et abaisse la concentration de la solution tant que celle-ci reste dans la zone métastable, entre courbes de saturation et de sursaturation.
L'extension du domaine métastable dépend d'effets cinétiques (vitesse de refroidissement, nombre de germes…) propres à chaque situation.
Solvatation dans l'eau
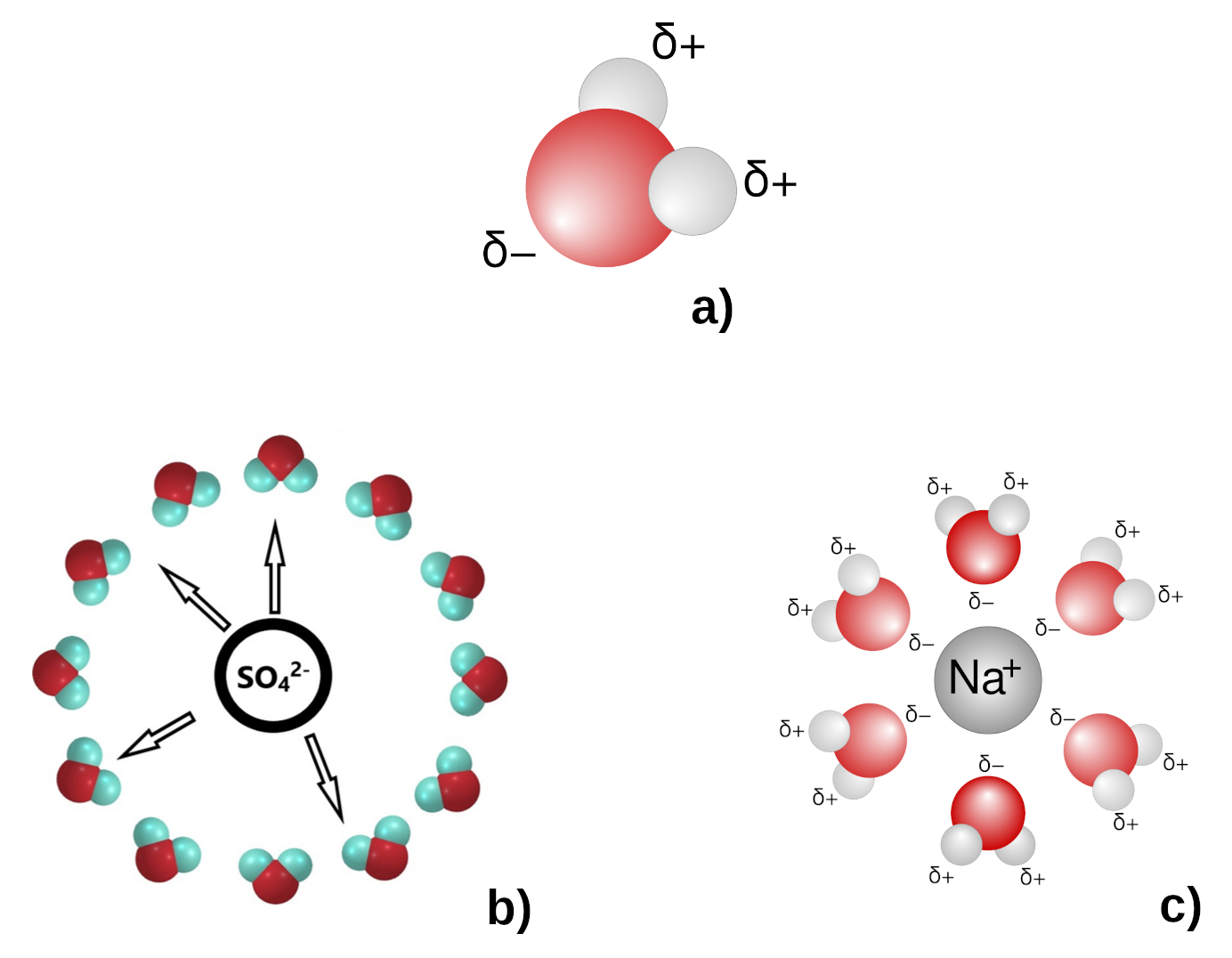
Lors de l'introduction d'un cristal dans un solvant, les atomes, ions ou molécules du cristal peuvent se disperser dans la solution et interagissent alors avec les molécules de solvant. Cette interaction s'appelle la solvatation. L'eau est un solvant polaire, c'est-à-dire que la position et les charges électriques des atomes d'oxygène et d'hydrogène forment un dipôle électrique (figure 8a). Le caractère polaire de l'eau facilite la mise en solution des ions.
Par exemple, les ions Cu2+ et SO42− sont solvatés. Le caractère polaire de la molécule d'eau permet d'associer les ions par couches enveloppantes afin d'assurer leur neutralité électrique (figure 8b). Des structures analogues peuvent se former autour des cations, mais avec les dipôles d'eau disposés dans le sens contraire (figure 8c).
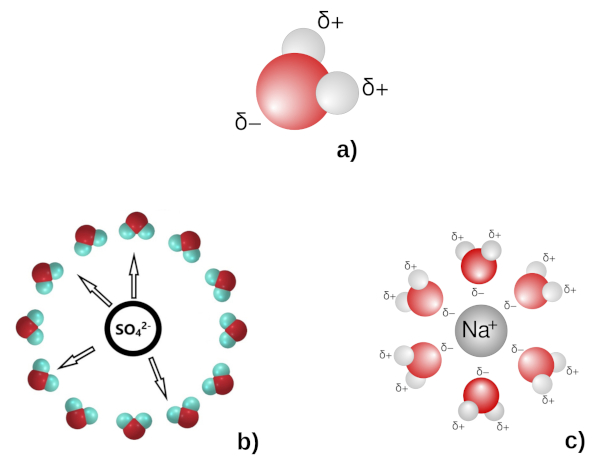
Source - © - — D'après wikimedia , 2006 (a et c) et Kulichenko et al., 2019 [ 16 ] (b)
a) L'eau est un solvant polaire (“excès” de charge négative côté oxygène et positive côté hydrogène).
b) Sphère de solvatation de SO42−, les hydrogènes “+” sont “tournés” vers le centre “négatif”.
c) Sphère de solvatation de Na+, les oxygènes “− ” sont “tournés” vers le centre “positif”.
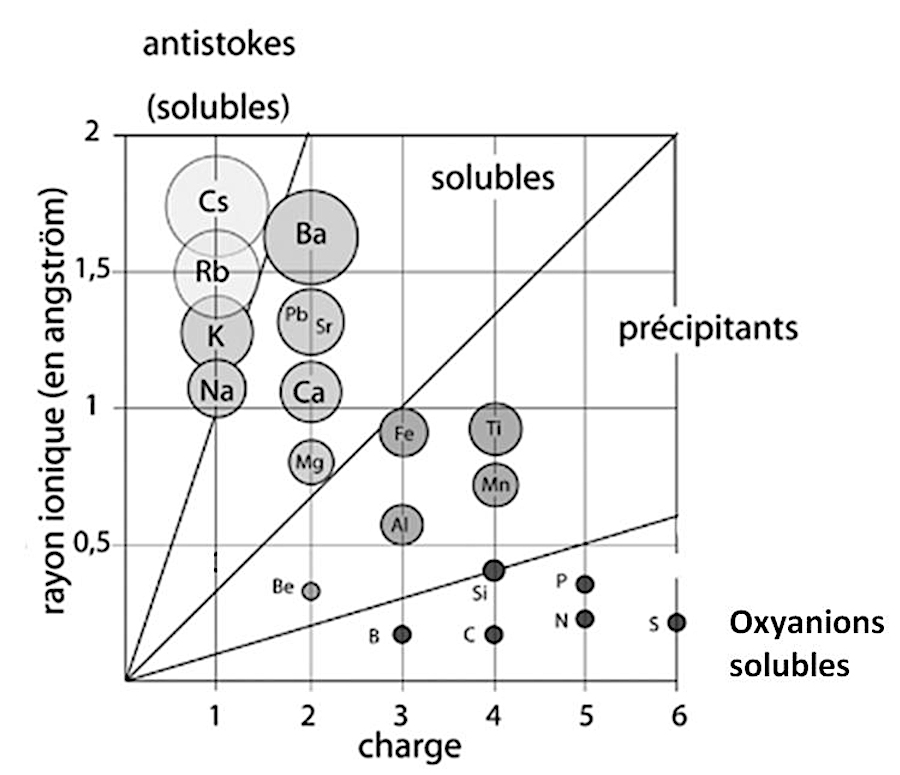
Un ion solvaté est entouré par des couches de molécules de solvant (eau). La sphère de solvatation est d'autant plus grande que l'ion est petit. Le volume de la sphère de solvatation de Na+ dans l'eau (volume efficace) est plus grand que celui de la sphère de solvatation de l'ion K+ (figure 9). Par ailleurs, plus la charge de l'ion est grande, plus il faut de dipôles d'eau pour la neutraliser.
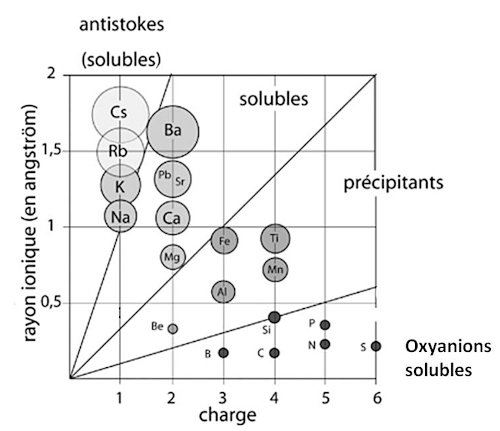
Source - © 2024 — Bernard Barailler
Par conséquent, en cas de compétition entre les ions, ceux qui ont le volume efficace (solvaté) le plus petit et la charge la plus faible vont rester en solution. Les autres auront tendance à précipiter. Le diagramme de Goldschmidt (figure 9) donne l'ordre de précipitation dans l'eau de certains cations : Fe2+, Ca2+, Na+, K+. On en verra les conséquences géologiques plus loin, dans le cas des évaporites.
Du soluté au cristal : une transition de phase dictée par la thermodynamique
Comme on l'a vu précédemment, la réaction chimique de dissolution/cristallisation est réversible : Cristal ↔ Soluté + Solvant.
Dans le cas de “Blue”, on a : CuSO4·5H2O (s) ↔ Cu2+ (aq) + SO42− (aq) + 5 H2O (l).
Pour savoir si la réaction penche vers la cristallisation ou la dissolution, il faut introduire les concepts de la thermodynamique qui nous indiquent que tout système naturel tend à minimiser son énergie afin d'atteindre l'état le plus stable.
Pour une réaction chimique générale ∑ α i R i ↔ ∑ β j P j ( réactants, produits, et les coefficients stœchiométriques de la réaction), il faut prendre en compte l'énergie libre de Gibbs G. La différence d'énergie libre (ΔG) entre les réactants Ri et les produits Pj est alors :
Δ G = Δ G 0 + RT ln ( Π a P j β j . a R i − α i ) = Δ G 0 + RT ln ( Q ), où est l'activité d'un composant.
À l'équilibre, il y a autant de cristallisation que de dissolution, donc Δ G éq = 0, ce qui donne Δ G 0 = − RT ln ( Π a P j β j . a R i − α i ) éq = − RT ln ( K sp ).
Donc Δ G = − RT ln ( K sp ) + RT ln ( Q ) = RT ln ( Q / K sp ), Q / K sp définissant l'index de saturation(lien externe - nouvelle fenêtre).
Par définition, la force motrice est : σ = Δ G / RT = ln ( Q / K sp ).
L'écart de l'index de saturation Q / K sp à la valeur 1, qui est la valeur d'équilibre, donne une mesure de la force motrice de la croissance ou de la dissolution à venir.
En résumé :
si σ = 0 (index de saturation = 1), le système est à l’équilibre, il n’est pas statique, il y a simplement autant de cristallisation que de dissolution,
si σ < 0 (index de saturation < 1), alors il y aura dissolution (la solution étant sous-saturée),
si σ > 0 (index de saturation > 1), alors il y aura cristallisation via les processus de nucléation et de croissance (la solution étant sursaturée).
Il s'agit, en termes de thermodynamique, d'une transition de phase. Lors de la cristallisation, on passe de l'état liquide (soluté) à l'état solide (cristal). L'état solide est alors plus stable, son énergie est plus basse que l'état initial en soluté. La différence d'énergie (Gcristal−Gsoluté<0) est négative, l'énergie est fournie par le système vers l'extérieur sous forme de chaleur. La réaction est exothermique. Pour continuer à croitre, le cristal doit évacuer la chaleur de sa surface où la réaction se produit vers le milieu extérieur. La chaleur s'évacue à travers le cristal et le fluide résiduel vers son environnement externe (parois en verre du bécher, épontes du filon…).
On voit donc que la croissance d'un cristal est conditionnée par deux facteurs essentiels :
l'apport d'unités de croissance et leur diffusion vers les faces du cristal,
l'évacuation de la chaleur, afin que la température à la surface n'augmente pas et que réaction exothermique de cristallisation perdure.
Les conditions de sursaturation d'une solution dépendent de la température (figure 4), de la pression, du type de solvant, du rapport entre la quantité de solvant et la quantité du minéral à mettre en solution, du pH, de la présence d'autres ions…
La figure suivante illustre l'incidence de la pression sur la courbe de solubilité. De façon imagée, plus la pression est forte plus on a des molécules de solvant proches pour solvater les solutés et donc limiter les interactions avec les autres molécules nécessaires à la cristallisation. Ainsi, à température donnée, la solubilité augmente avec la pression.
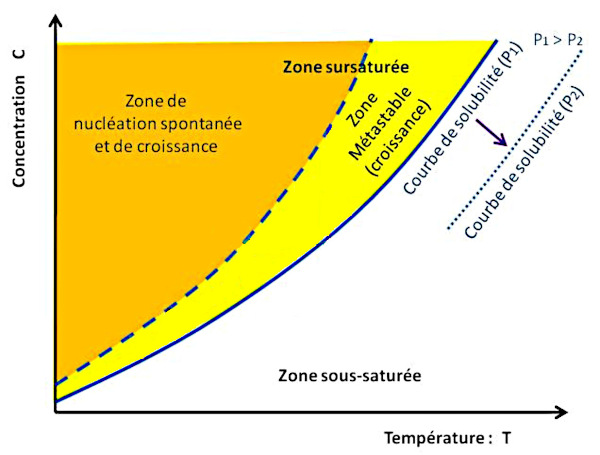
Source - © 2024 — Bernard Barailler
Une genèse par étapes : nucléation, croissance, murissement
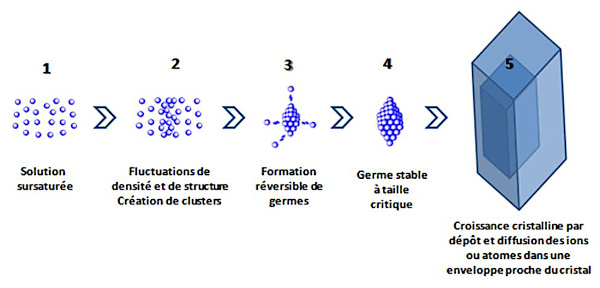
Source - © 2024 — Bernard Barailler
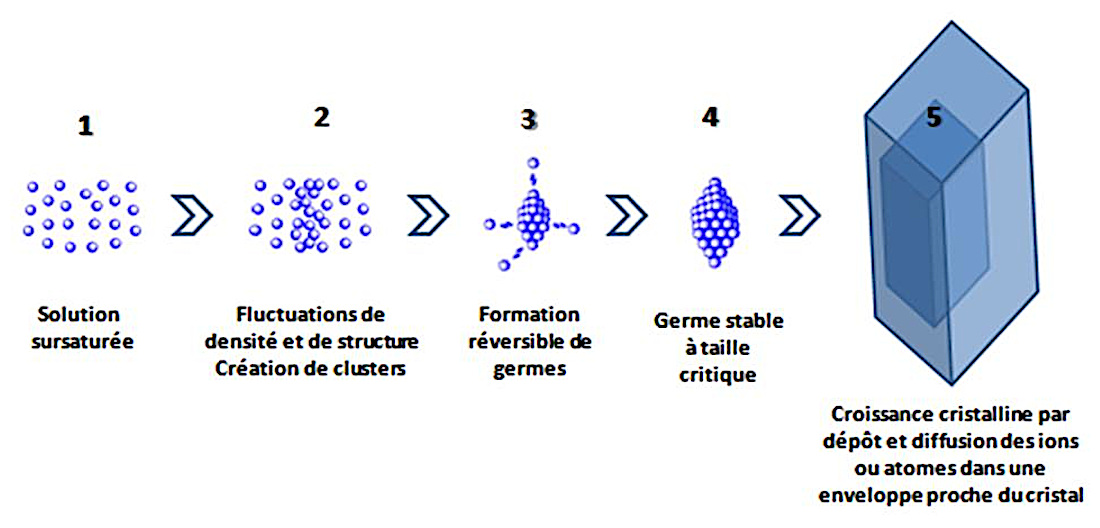
Un cristal se forme en plusieurs étapes (figure 11). Dans une solution sursaturée (1), les ions et les molécules sont animés de mouvement liés à l'agitation thermique.
Lors des rencontres des espèces chimiques, il peut se produire des fluctuations de densité (2). Dans la solution, en fonction du degré de saturation, des agrégats peuvent commencer à s'auto-organiser au gré de ces fluctuations de densité. Ces agrégats, ou clusters, comprennent quelques atomes ou quelques molécules. Ils se sont constitués par une série de réactions bimoléculaires entre molécules ou ions du soluté (Espitalier et al., 2024 [13]). Ils peuvent s'assembler, mais aussi se détruire, au gré de ces fluctuations.
Toutefois, au delà d'une certaine énergie d'activation, des germes de cristaux peuvent se construire à partir des agrégats (3). Les étapes (2) et (3) concernent la nucléation, c'est-à-dire la fabrication de germes. Ces germes ne sont pas stables en dessous d'une certaine taille critique, ils peuvent se dissoudre à nouveau dans le solvant. Durant cette phase, la préexistence de cristaux de même nature peut favoriser une nucléation secondaire de contact. Par exemple lors de la circulation d'un fluide hydrothermal chargé en silice (voir plus loin) les cristaux de quartz peuvent se développer sur les phénocristaux de quartz de l'encaissant granitique. De même une circulation fluide chargée en ions carbonate et calcium peut, dans une fente de calcaire, déposer ces cristaux sur les microcristaux de calcite constituant ce calcaire.
À partir d'une certaine taille critique, les germes deviennent stables. La croissance cristalline s'installe à partir d'un germe stable (4).
La croissance d'un cristal s'opère par la superposition de surfaces, couche après couche (5). Ces couches sont formées à partir d'unités de croissance (ions, molécules…).
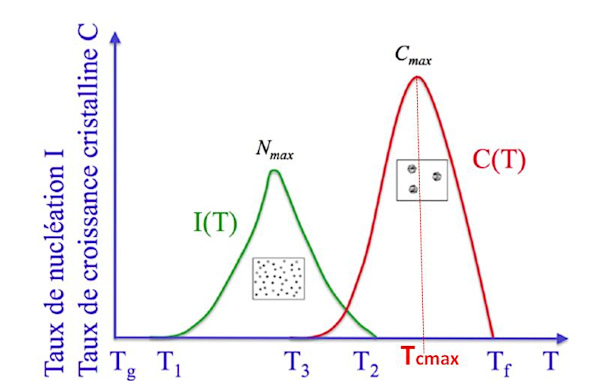
Source - © 2024 — Bernard Barailler
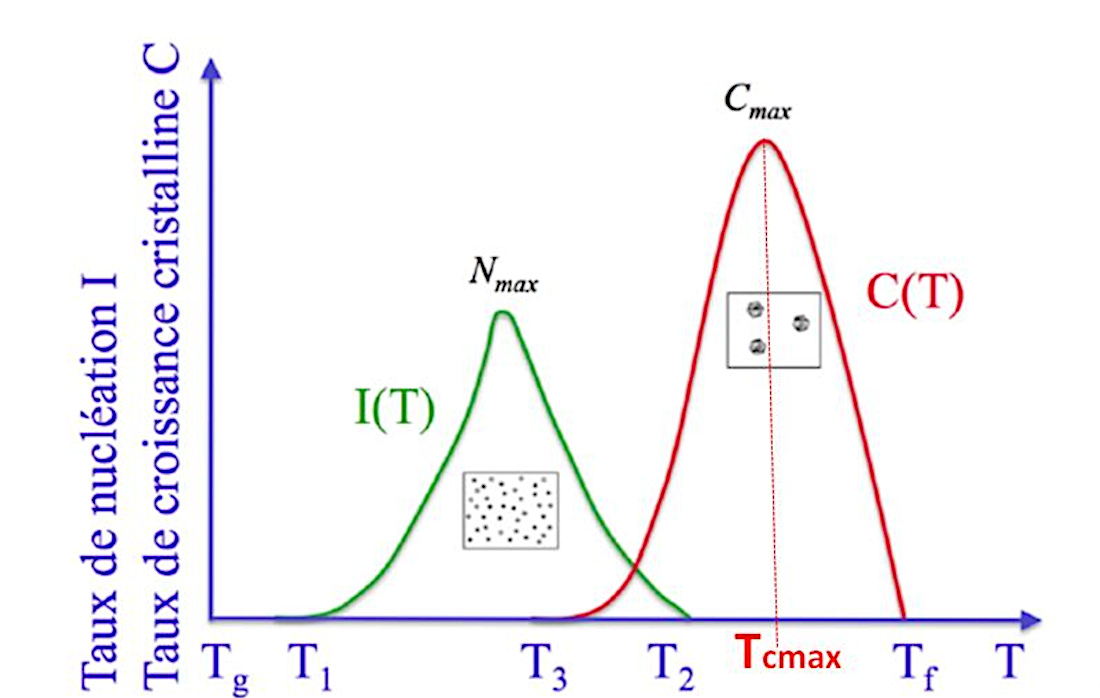
La figure 12 montre que, pour un cristal donné, la température T3 à laquelle le taux de nucléation (formation des germes) est maximal, est inférieure à la température Tcmax à laquelle la vitesse de croissance du cristal est maximale.
En effet, si la température est élevée, l'agitation thermique l'est également par définition. Les chocs des ions et des molécules sont donc plus forts et plus fréquents et les germes ont du mal à y résister (rappel : la réaction est réversible). Par ailleurs, pour la croissance cristalline, la diffusion des unités de croissance est meilleure, donc les cristaux croissent “mieux” (jusqu'à une certaine limite de température).
Ainsi, si on veut faire croitre rapidement un cristal à partir d'un germe existant, on a intérêt à se maintenir vers cette température critique Tcmax de croissance maximale.
Enfin, un dernier phénomène, appelé murissement d'Ostwald, conduit à l'augmentation de la taille moyenne des cristaux issus de la solution. Quand des cristaux sont en suspension dans une solution dont la sursaturation devient faible, les plus petits d'entre eux ont tendance à se dissoudre à nouveau. Lorsqu'on fait des allers-retours en température, on va progressivement faire disparaitre les petits cristaux au détriment des plus gros (pour une même durée de dissolution, les plus petits cristaux ont une probabilité plus importante de totalement disparaitre, car ayant moins de matière et un rapport surface/volume plus grand – plus grande surface de contact et donc d’échange avec le soluté par unité de volume de cristal).
L'apport des unités de croissance pour continuer la réaction chimique de transition de phase est essentiel à la formation d'un cristal.
La croissance d'un cristal est souvent une activité cyclique. En effet, si on combine les effets des variations de concentration et de température, on voit qu'à partir d'une solution “chaude” dans le domaine de solubilité, une baisse de température va permettre d'atteindre la zone de nucléation. La croissance de cristaux diminue alors la concentration de la solution et peut augmenter la température (réaction généralement exothermique), au moins “localement” autour des cristaux, ce qui peut faire sortir du domaine métastable et donc aboutir à un arrêt de la cristallisation. Le retour de la solution dans le domaine métastable – où la croissance des cristaux existants est possible – fera repartir cette croissance, soit suite à une baisse de température (refroidissement général du système et/ou dispersion de l'énergie thermique localement générée par la cristallisation), soit par une augmentation de la concentration (soit locale par réhomogénéisation de la solution appauvrie autour des sites de cristallisation par simple diffusion des éléments en solution, soit générale par ajout de soluté ou départ de solvant – évaporation, par exemple).
Expérimentalement, à la température de croissance maximale Tcmax, on peut maintenir le niveau de concentration par apport de soluté et on peut assurer un flux des unités de croissance avec une convection forcée adaptée (sauf si on veut privilégier un seul cristal comme dans le cas de Blue).
Dans la nature, A. Abréal (2010 [2]) décrit une cristallisation des grenats dans les pegmatites (filons à refroidissement rapide) par étapes. Des phases de baisse de température avec perte de solvant – et donc augmentation de la concentration – amènent à la cristallisation (trajets en diagonale sur la figure 13 ci-dessous), des phases de cristallisation rapide à température constante aboutissent à un arrêt de cristallisation par baisse de concentration (“descentes” verticales sur la figure 13). La température du système continuant à baisser, un retour à la cristallisation s'ensuit.
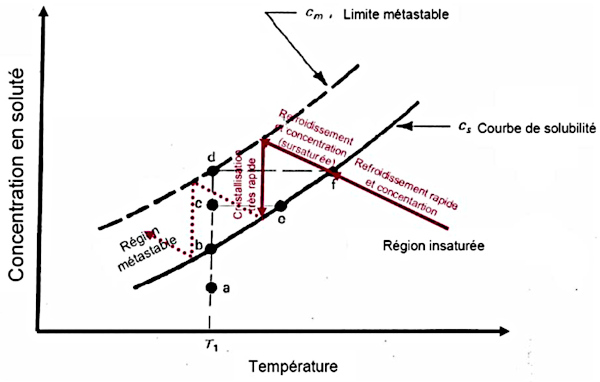
Source - © 2010 — D’après Abréal [ 2 ] , modifié
Dans un système globalement en baisse de température, des phases de cristallisation rapide avec forte baisse de concentration aboutissent à des arrêts temporaires de cristallisation.
Jusqu'à présent nous n'avons considéré que la modélisation thermodynamique de manière simplifiée. Mais une considération purement thermodynamique ne permet pas de savoir si la précipitation “possible” va mettre quelques millions d'années ou quelques secondes. Les aspects cinétiques et l'énergie d'activation (énergie thermique qu'il faut apporter pour qu'une réaction chimique possible ait réellement lieu) sont d'autres paramètres à considérer.
En effet, pour cristalliser à partir d'un fluide, il faut opérer une transition de phase : passage de la phase liquide à la phase solide. Souvent, ceci est modélisé par le fait qu'un cristal stable a la plus faible énergie libre de Gibbs, notée G. Mais cela suppose en général un système fermé isolé, à pression P et température T constantes, sans apport supplémentaire de matière. Et on se place aussi dans une hypothèse de transition réversible (quasi-statique).
Or, dans la nature, il y a souvent un gradient de température, lié aux différentes sources de chaleur dans la Terre. La pression est également susceptible d'évoluer notablement, notamment avec les phénomènes géodynamiques. On est en système ouvert, des apports de matière s'opèrent par convection, diffusion, advection… Des éléments chimiques nouveaux peuvent rentrer en jeu (impuretés). Les conditions aux limites du système hydrothermal sont très variables : filon, karst, géode, pli… Bref, on est loin de la thermodynamique réversible d'un système fermé, isolé du monde extérieur. On est plutôt dans la thermodynamique des systèmes irréversibles non linéaires avec des conditions aux limites complexes.
Appliquons les considérations théoriques ci-dessus à des exemples naturels pour nous aider à comprendre comment certains cristaux se développent dans la nature.
Des exemples avec de l'eau “de surface”
Gypse, la genèse d'une rose des sables
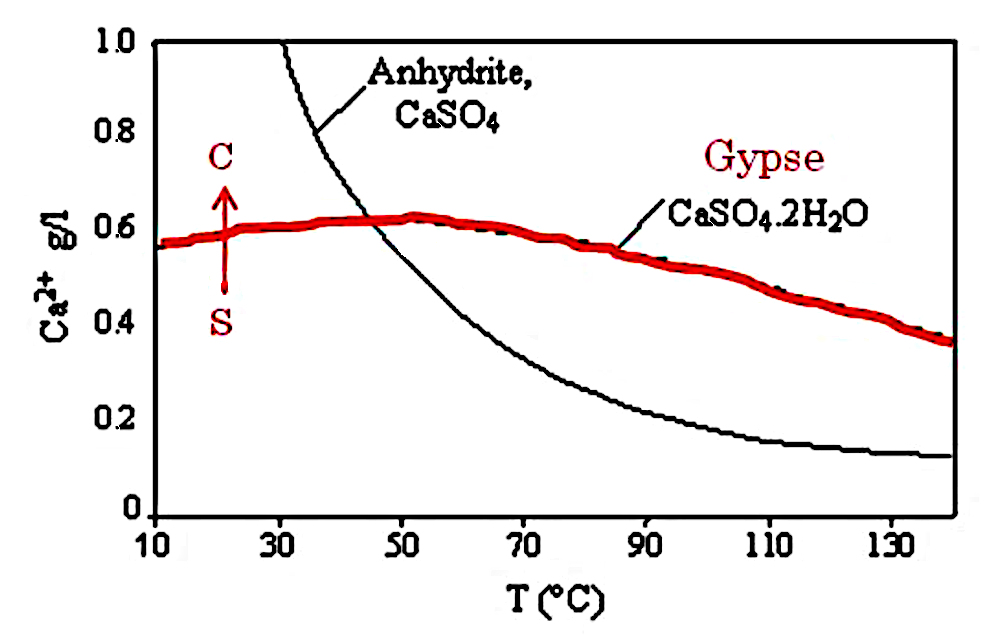
La solubilité du gypse et celle de l’anhydrite sont données dans la figure suivante.
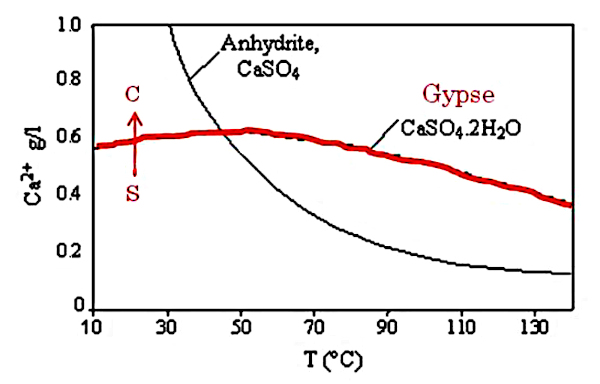
Source - © 2003 — D'après Pelser at al. [ 18 ] , modifié
À température et pression ambiantes, on voit que le gypse peut cristalliser par évaporation en suivant le trajet de S (soluté) vers C (cristal).
Dans quelles conditions géologiques la cristallisation de gypse se produit-elle ?
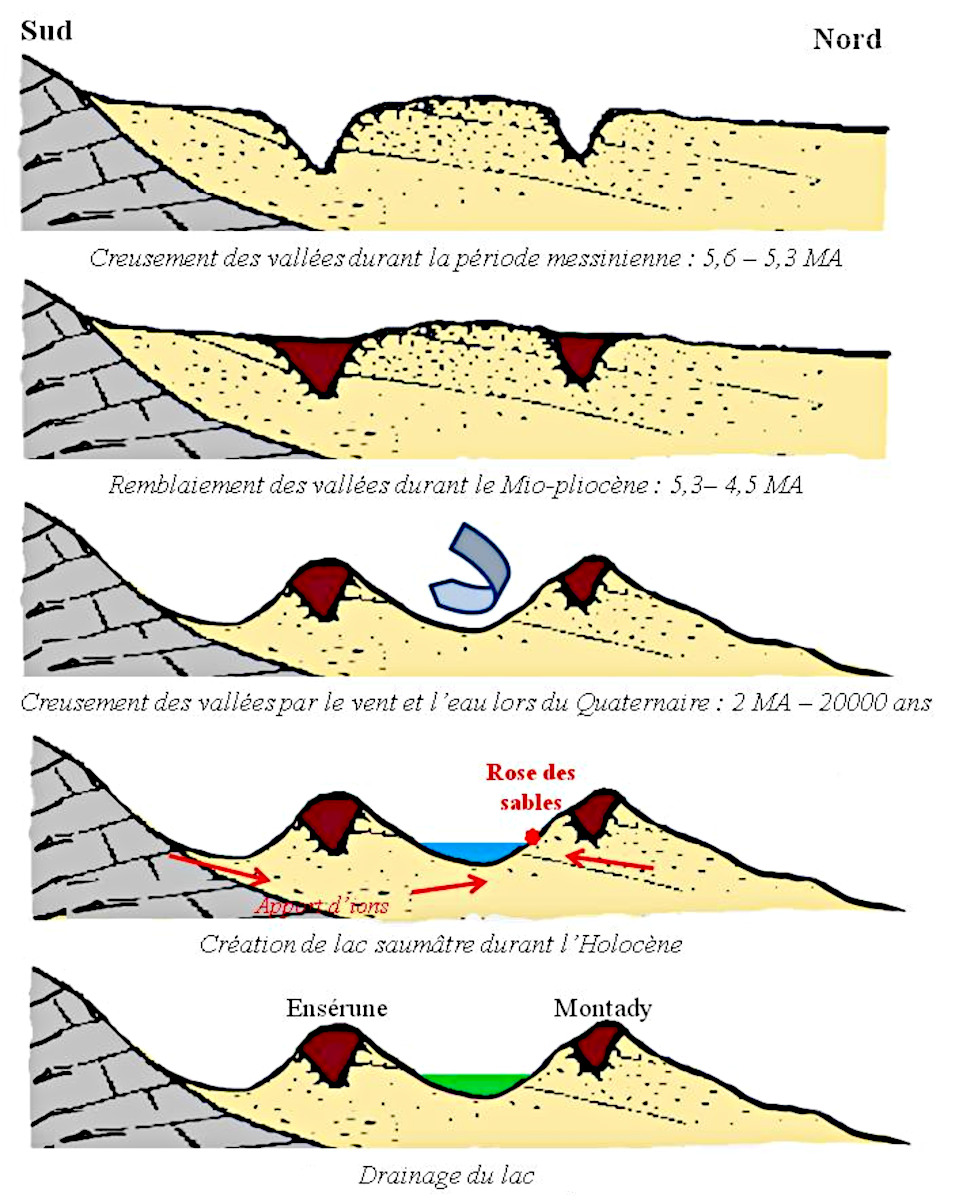
Prenons l'exemple du bassin de Montady dans l'Hérault (cf. P. Thomas, 2009 [30]). Il y a 5,6 millions d'années, lors de la crise messinienne (fin du Miocène), le niveau de la mer était alors beaucoup plus bas qu’à l’actuel (et que précédemment). Les rivières ont retrouvé leur profil d’équilibre en incisant la roche (sables miocènes), creusant des vallées, qui ont ensuite été remblayées durant la période suivante du Mio-Pliocène. Au Quaternaire, le lac se remplit entre Montady et Ensérune. Durant la période quaternaire du Greenlandien (−11,7 à −8,2 milliers d'années) les contrastes saisonniers sont très marqués. À cette période, un climat sec semi-aride était probablement à l'œuvre (Abbé et al., 2009 [1], figure 15).
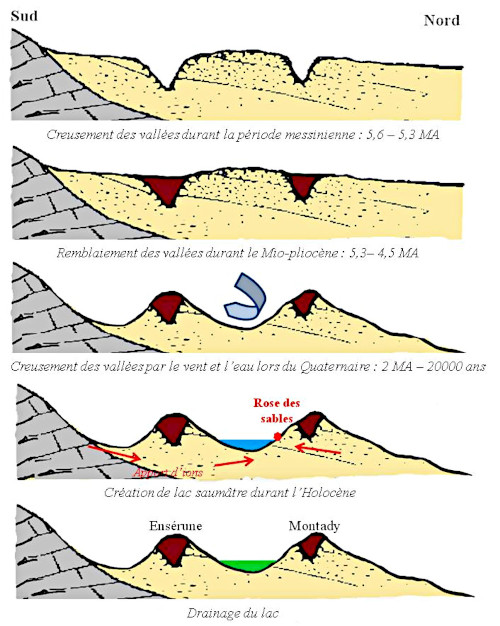
Source - © 2009 — D’après Abbé et al. 2009 [ 1 ]
Calcaires en gris, argiles et sables du Miocène en jaune, remblaiement mio-pliocène en rouge-brun, lac saumâtre holocène en bleu, lac drainé en vert.
À l'Holocène, le niveau de la nappe qui alimente l'étang de Montady fluctue au gré des saisons. Au toit de la nappe, l'eau chargée d'ions calcium Ca2+ et sulfate SO42− percole et s'infiltre dans les argiles et les sables du Miocène. Les ions calcium peuvent provenir de la dissolution par les eaux de pluie des massifs calcaires environnants. En effet l'eau de pluie chargée en dioxyde de carbone CO2 se transforme en un acide faible.
2 H2O + CO2 → H2CO3 + H2O ↔ H3O+ + HCO3− ; HCO3− est l'ion hydrogénocarbonate.
Cet acide en contact avec la calcite, libère l'ion Ca2+ selon la réaction :
HCO3– + CaCO3 + H2O ↔ Ca2+ + H3O+ + 2 CO32−.
Les ions sulfates, SO42−, peuvent provenir, par exemple, d'un biseau salé de la mer proche ou du lessivage de sédiments marno-calcaires de l'Oligocène.
Avec l'évaporation, la concentration des ions augmente (trajet de S vers C, figure 14). Ceux-ci précipitent alors pour donner les évaporites dans la partie aval émergé du lac (ou chott[2] ). Les ions se déposent sur les faces d'un germe. Il s'agit d'une activité cyclique avec un apport d'unités de croissance progressif (figure 17).
Au gré des percolations successives, les cristaux de gypse se développent en écartant progressivement l'encaissant meuble (sables argileux). Le milieu extérieur de croissance étant relativement isotrope (sable mouillé), il n'y a pas d'axe de croissance privilégié. Les cristaux se développent alors dans toutes les directions et sont de forme lenticulaire. En effet, la croissance des faces est gênée, les grains de sable mêlés d’argiles freinent la diffusion des unités de croissance, dont l’accrochage s’effectuera préférentiellement au niveau des arêtes, d'où une forme aplatie, lenticulaire (voir modèle de Kossel – Kern, 1968 [15] – et théorie BCF – Burton et al., 1951 [9]). Les cristaux forment alors des macles et des associations en pétales de rose (Soleilhavoup, 2011 [23]), Dan et Yaalon 1982 [11]) (figure 16). De là vient le nom de rose des sables (Abbé et al., 2009 [1]).
À Montady, on obtient finalement des roses greenlandiennes des sables miocènes.
 Source - © 2024 — Bernard Barailler Voir aussi Comment se forment les roses des sables ?. Rose des sables d’environ 6 cm de diamètre. |
 Source - © 2024 — Bernard Barailler Les stries de croissance attestent de la cyclicité du phénomène. Taille de l’échantillon environ 5 cm. |
Les évaporites
Les évaporites sont des roches sédimentaires formées par précipitation de sels à partir d'une masse d'eau qui se concentre par évaporation.
Il y a 5,6 Ma, lors de la crise messinienne, la Méditerranée se retrouve pratiquement isolée de l’océan Atlantique (Dromart, 2020 [12]). En effet, la croissance de la calotte polaire antarctique abaisse de façon importante le niveau marin général. Par ailleurs, des mouvements tectoniques liés à la convergence Afrique-Europe réduisent voire ferment le corridor de Gibraltar (Martìn et al., 2002 [17]). La Méditerranée s’est alors retrouvée sous-alimentée. L’évaporation y domine alors les apports d’eaux douce et océanique. La concentration en sels augmente progressivement, puis elle franchit les courbes de solubilité associées et provoque des dépôts de sels dans ses deux grands sous-bassins. Cette crise et cette chute importante du niveau de la Méditerranée est aussi connue pour le fort surcreusement des vallées fluviales péri-méditerranéennes (les rias messiniennes) (Dromart, 2020 [12]).
La formation d'évaporites à partir d'eau de mer est un phénomène bien reconnu dans le passé et sur le pourtour méditerranéen actuel (Thomas, 2015 [31], 2018 [32]). Notons que des dépôts équivalents se forment aussi en contexte continental à partir d'eaux de ruissellement ou d'eaux hydrothermales (Schultz, 2015 [24], Thomas, 2019 [33]).
En 1971, Douglas James Shearman [22] réalise l'expérience suivante : il fait évaporer de l'eau de mer à la pression atmosphérique. Il constate alors la séquence de cristallisation suivante :
hydroxydes de fer, calcite CaCO3, dolomie CaMg(CO3)2 ;
gypse CaSO4·2H2O, anhydrite CaSO4 ;
halite NaCl ;
carnallite KMgCl3·6H2O et polyhalite K2MgCa2(SO4)4·2H2O ;
sylvite KCl.
Cette séquence peut s'imager, en simplifiant, par le fait que les ions Ca2+ ont besoin de plus de molécules d'eau autour d'eux pour être solvatés que les ions Na+ ou K+ (cf. conclusion de la partie Solvatation dans l'eau). Les ions sont en compétition les uns avec les autres pour avoir des molécules d'eau autour d'eux leur permettant de rester solvatés. Ceux qui en ont moins besoin (K+, ici) précipitent en dernier.
Évaporites et géométrie de dépôt
Dans le cas d'un bassin fermé dans laquelle une saumure s’évapore, les dépôts pourront former des cercles concentriques (en carte) qui correspondent aux dépôts progressifs : les dépôts successifs ne se réalisent que vers le centre du bassin, les bordures étant petit à petit exondées du fait de l'évaporation et de la baisse du niveau d'eau.
L’évaporation théorique d’une colonne de 1000 m d’eau de mer (à 34,5 g/L de sels) aboutirait à une quinzaine de mètres de dépôt d’évaporites, à savoir, par ordre de dépôt, 0,05 m de carbonates, 0,5 m de gypse, 12 m de halite, et 2,6 m de sels de K et Mg (Rouchy et Blanc-Valleron, 2006 [21]).
Dans la nature, des géométries variées sont possibles dès lors que se mettent en place des gradients de concentration dus à une alimentation continue ou intermittente en eau de mer et/ou en eau douce ou plus ou moins concentrée, à proximité de la mer ou en milieu purement continental (figure ci-dessous pour un bassin alimenté en eau de mer). La taille, la géométrie et la profondeur du bassin contrôlent aussi la géométrie des dépôts évaporitiques.
Attention aux schémas trop simples (et trop répandus sur le web et dans bien des livres) dans lesquels les évaporites sont représentées en coupe par une couche unique dont la nature change latéralement (carbonates aux bords et potasse au centre) ! Il y a bien dépôt successif de couches de natures différentes et d’extensions latérales variables, qui se superposent (même si on n’a pas, localement, la totalité de la série évaporitique sur une verticale – dans un forage).
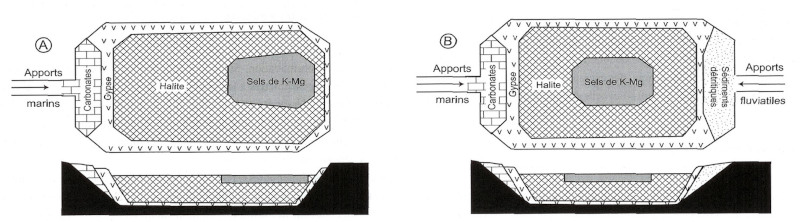
Source - © 2006 — D’après Rouchy et Blanc-Valleron [ 21 ]
Schémas, en carte et en coupe, de la distribution des dépôts évaporitiques dans un bassin alimenté uniquement par de l’eau de mer (A) ou par eau de mer et eau douce (B).
La calcite CaCO3 et le dioxyde de carbone CO2
Un autre exemple classique dans la nature est la précipitation de la calcite à partir d'ions calcium Ca2+ et d'ions hydrogénocarbonates HCO32− issus de la dissolution de CO2 dans l'eau.
Ca2+ + 2 HCO3− ⇌ CaCO3(s) + CO2(g) + H2O
Pour que la calcite précipite, il faut que [Ca2+].[HCO3−]2 / PCO2> Keq, avec Keq = [Ca2+]eq.[HCO3−]eq2/ PCO2éq la constante d'équilibre de la réaction. C'est le phénomène de sursaturation. On voit que si la pression partielle de CO2 est grande, la calcite aura plus de mal à précipiter. À l'inverse lors d'un dégazage de CO2, la calcite aura tendance à précipiter et cristalliser.
D'autre part, la solubilité du CO2 dans l'eau augmente avec la pression et diminue lorsque la température augmente. Ainsi, si l'air d'une grotte est plus chaud que l'eau qui suinte au plafond et que la pression partielle de CO2 y est faible, alors on va avoir dégazage de CO2 et formation de stalactites et de stalagmites par précipitation de calcite (figure 18). De la même façon, si un fluide remonte le long d'une fissure, on aura une baisse de pression et on pourra avoir une cristallisation de filons de calcite (Thomas, 2021(a) [34]).

Source - © 2024 — Bernard Barailler
Longueur de l’échantillon d’environ 8 cm.
Des exemples avec de l'eau supercritique
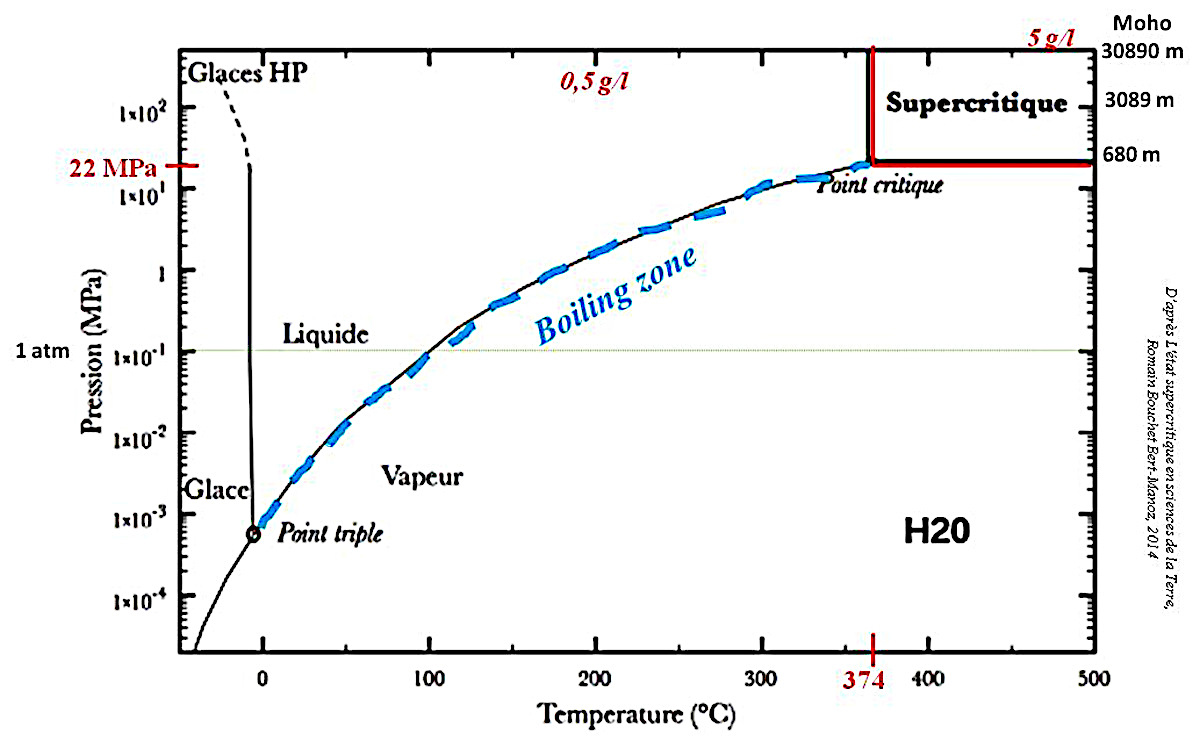
Les eaux de surface et les “superpouvoirs” de l'eau supercritique
La Terre est la planète bleue. L'eau est le solvant le plus fréquent dans la nature. On pense généralement à l'eau de pluie, eau que l'on retrouve dans les eaux continentales, plus ou moins chargée en éléments dissouts selon son parcours (surface, nappes) et les formations géologiques rencontrées. L'eau de mer résulte, elle, d'une concentration des eaux continentales et de précipitations au sein des océans et d'échanges avec la lithosphère océanique. D'autres origines de l’eau sont possibles. De l'eau peut provenir d'une activité volcanique ou de processus de métamorphisme libérant des fluides. Surtout, ces eaux peuvent circuler dans des formations géologiques variées (bassins sédimentaires, karsts…) et y séjourner plus ou moins longtemps. On a donc des eaux de propriétés différentes qui vont acquérir des compositions chimiques très variées, indicateurs possibles des conditions de genèses de certains cristaux (cf., par exemple, les calcites noires et blanches dans les septarias – Barailler, 2023(b) [6]).
Un autre facteur important est l'état physique de l'eau. Pour une pression supérieure à 22,1 MPa et une température supérieure à 374°C, l'eau est dans un état dit supercritique qui lui confère des “superpouvoirs” de solubilisation (Bouchet Bert-Manoz, 2014 [8]). La solubilité dans de l’eau supercritique suit la courbe d’évolution continue en fonction de P et T décrite dans l’eau liquide, il n’y a pas de saut de solubilité (concentration maximale en solution), mais modification des propriétés du fluide. La tension superficielle disparait, l’eau se comporte alors plus comme un gaz et peut s’insérer facilement dans le moindre interstices, mouiller la surface des cristaux, et faire diffuser rapidement les éléments présents en solution. Ainsi, une eau supercritique dissout, par exemple, sulfures et silice plus “facilement” (du fait de P, T et de l’état supercritique) et donc en plus grande quantité qu'une eau liquide de surface qui circulerait au contact des mêmes roches. Ces eaux supercritiques “chargées” vont ensuite se “décharger” lorsqu'elles vont quitter le domaine supercritique, généralement en remontant vers la surface.
Le quartz

Source - © 2024 — Montage Bernard Barailler
La silice est soluble dans l'eau, heureusement pour nous, en très petite quantité, sinon on aurait des problèmes pour boire de l'eau dans nos verres. À la pression atmosphérique, la limite de solubilité dans l’eau est de l'ordre de 120 mg·L−1 à 25°C (Rimstidt, 1997 [20] ; Alexander et al., 1954 [3]), la silice amorphe étant légèrement plus soluble que le quartz (cristallisé).
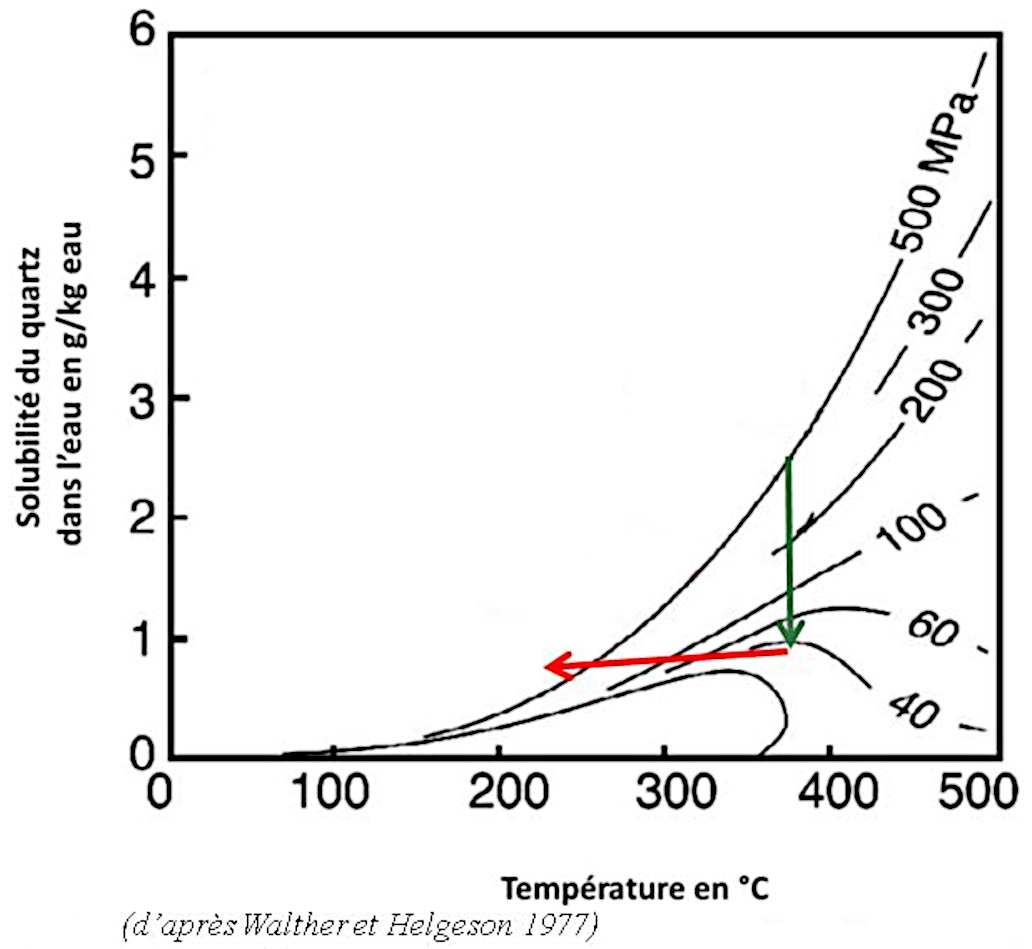
La silice dissoute est sous forme d'acide orthosilicique H4SiO4. La figure suivante (figure 20) montre la solubilité du quartz dans l'eau en fonction de la température et de la pression. À 200 °C et 200 MPa, conditions rencontrées vers environ 6 km de profondeur, on peut dissoudre de l’ordre de 500 mg de SiO2 en solution dans 1 kg d'eau.
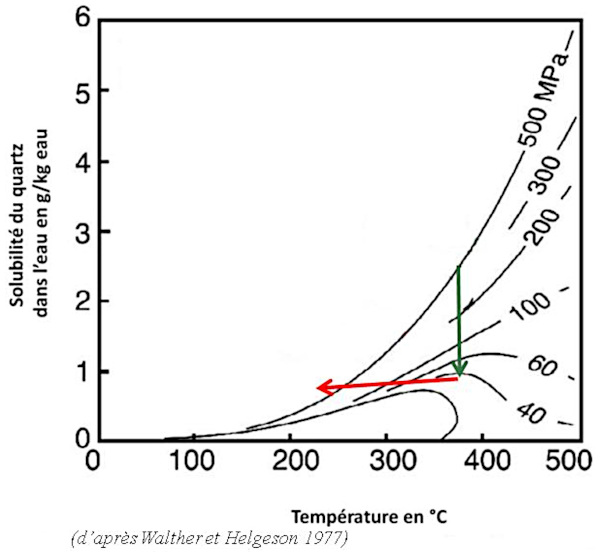
Source - © 1997 — D'après Walther et Helgeson [ 36 ] , modifié
Lorsque la pression baisse (trajet vert) ou que la température baisse (trajet rouge), la solubilité du quartz baisse assez rapidement.
Quand un fluide remonte le long de failles ou de fractures, la pression baisse fortement. Lorsque les échanges thermiques avec les épontes se font dans la durée, la température baisse également pour se mettre à l'équilibre thermodynamique. Les conditions de cristallisation sont alors réunies.
L'eau acquiert des “superpouvoirs” dans deux zones bien définies : l’eau supercritique et dans la “boiling zone” (figure 21).
Tout d'abord, l'eau supercritique (T > 374°C et P > 22,1 MPa) peut contenir jusqu'à 5 g de SiO2 dissoute par kg d'eau (Fournier et Potter, 1982 [14] ; Rendel et Mountain, 2023 [19]). D'autre part, dans la “boiling zone” (à la limite entre domaine liquide et domaine vapeur – figure 21), il y a coexistence des phases vapeur et liquide. Le “bouillonnement” des fluides dans les fractures peut modifier fortement la vitesse de cristallisation, car, à température élevée, les transferts par la phase gazeuse de chaleur (évacuation de la chaleur de cristallisation) et d'unités de croissance sont alors facilités.
Un fluide supercritique a une forte capacité de solvatation et d'extraction, qui est due à sa faible viscosité et à sa forte diffusivité. Les fluides supercritiques – H2O mais aussi CO2, méthane… – qui circulent dans les roches sont capables d’atteindre et de mettre en solution un grand nombre d'éléments chimiques (silicium, soufre…).
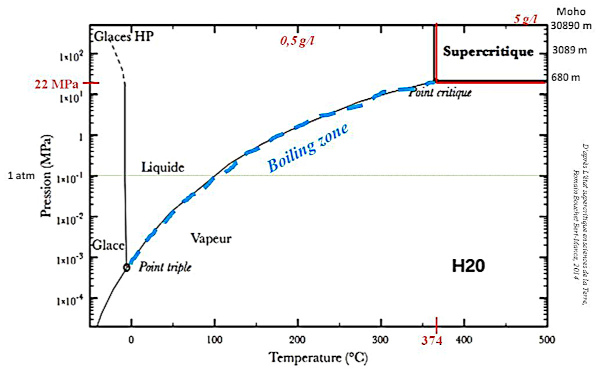
Source - © 2014 — D'après Bouchet Bert-Manoz [ 8 ] , modifié
La solubilité de la silice atteint 5 g/L en zone supercritique, contre seulement 0,5 g/L à 200 MPa et 200°C.
Les conditions d'obtention de l'eau supercritique sont nombreuses mais représentent des contextes géologiques particuliers.
Par exemple, l'eau circulant dans les fractures de la croute océanique à proximité des dorsales peut se trouver à l'état supercritique car P>22 MPa dès que la profondeur est supérieure à environ 2200 m, et on arrive à T>374°C à des profondeurs relativement faibles dans ces zones de fort gradient géothermique (remontée de l'asthénosphère et de l'isotherme 1200°C).
Dans les zones de subduction, l'antigorite (variété de serpentine – (Mg,FeII)3Si2O5(OH)4) se déshydrate à une pression de 2 000 MPa bien supérieure à la pression critique. La température est alors de l'ordre de 600-700°C : l'eau libérée par la déshydratation est alors supercritique.
Quand l'eau circule dans la croute continentale, elle peut être rapidement à l'état supercritique. La température critique est atteinte vers 12 km de profondeur (pour un gradient géothermique crustal moyen de 30°C/km), la pression critique, elle, dès 2,2 km de profondeur en pression hydrostatique (eau "en connexion" avec la surface) et même à plus faible profondeur, vers 700-800 m, dans le cas d'une eau soumise à la pression lithostatique.
Les zones de collision (Alpes, Pyrénées…) sont propices aux formations de filons hydrothermaux. Un métamorphisme de haute pression et haute température (HP-HT) produit la déshydratation de minéraux. Cette eau libérée est alors supercritique et peut dissoudre des éléments chimiques en grande quantité.
On a donc de nombreuses conditions géologiques propices à ce que des fluides avec de l'eau supercritique, fortement "chargés", remontent dans la croute. Quand la pression et/ou la température de ces fluides repassent sous les valeurs critiques, ils deviennent sursaturés. Les silicates et les sulfures peuvent alors cristalliser dans la croute supérieure sous forme de filons (dits hydrothermaux).
 Source - © 2021 — Pierre Thomas [ 35 ] Le quartz le plus grand mesure 6 cm de longueur. |
 Source - © 2024 — Bernard Barailler Quartz d’une longueur de 17 cm. |
La courbe présentée à la figure 21 correspond à de l'eau pure. D'autres paramètres importants sont parfois à prendre en compte. Par exemple, l'ajout de CO2, qui est un non-électrolyte, tend toujours à dilater le liquide ; il diminue alors l'hydratation et donc la solubilité, par exemple, de la silice. À contrario, aux faibles densités d'eau, l'ajout de sel NaCl provoque une contraction notable du volume de liquide et réduit la distance moyenne entre les molécules en interaction. Ceci améliore l'hydratation et augmente ainsi la solubilité, par exemple, de cette même silice. Notons, que la présence de saumures dans les fluides est fréquente dans les Alpes car ces derniers ont pu traverser les évaporites sous-jacentes du Trias.
Les fluides peuvent aussi être radioactifs et participer à la coloration des quartz en les rendant fumés (Barailler, 2024 [7]). Ces fluides peuvent aussi se refroidir très rapidement et donner une structure amorphe de la silice : l'opale hyalite (Barailler, 2023(a) [5]), voire former des dépôts singuliers comme des quartz dans des phryganes fossiles (figure 65 de Thomas, 2008 [29]). Notons aussi l’existence d’eaux hydrothermales riches en silice qui précipitent de la silice plus ou moins hydratée à leur arrivée en surface (Thomas, 2005b [27], 2005c [28]).
Enfin, on peut opérer la croissance contrôlée de quartz par la méthode dite de synthèse hydrothermale dans des autoclaves (figure 23) (Thomas, 2005a [26] ; Zaccaro et Ibanez, 2014 [37]). Ces cristaux sont utilisés dans l'industrie, en électronique numérique pour ses propriétés piézo-électriques (montres à quartz, oscillateurs, capteurs…) et pour ses propriétés optiques (prismes, lentilles) ou encore en verrerie de pointe.
Dans les secrets des filons
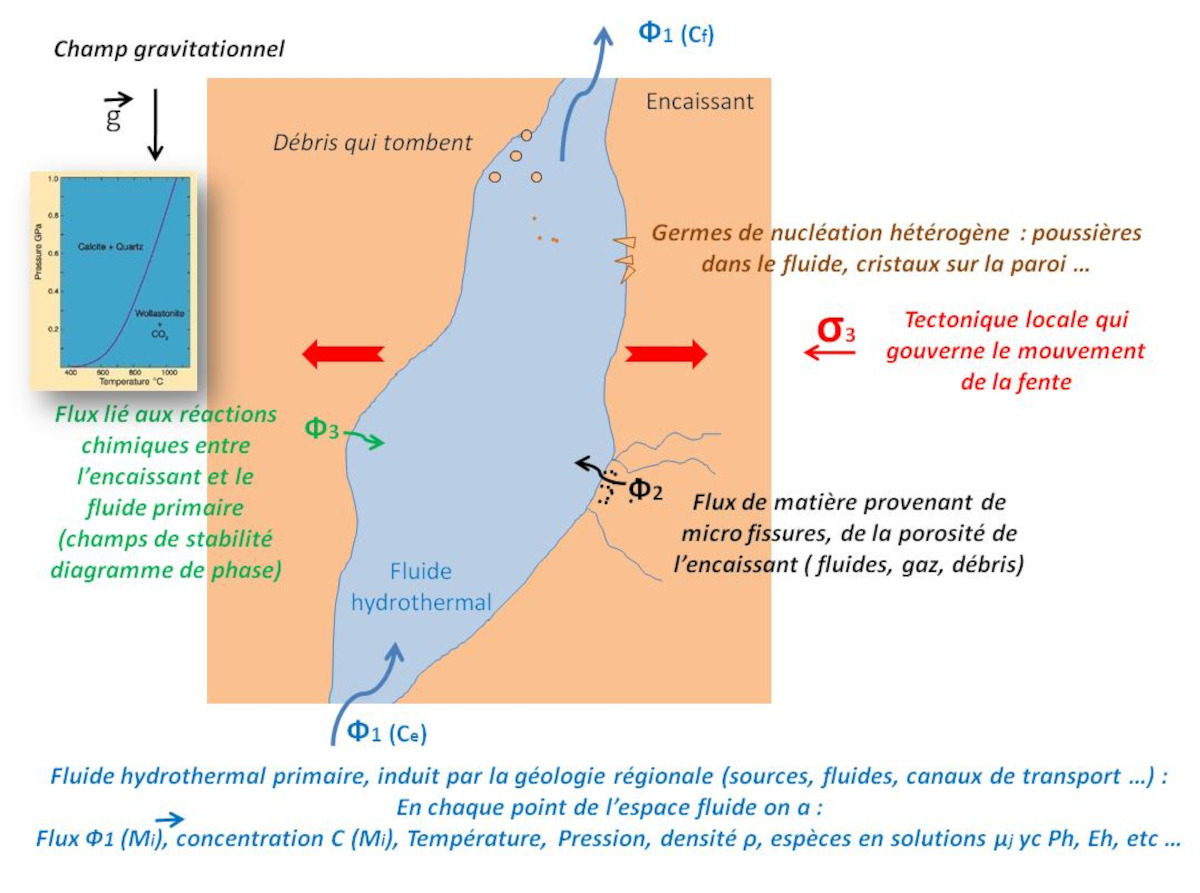
Mais que se passe-t-il précisément dans un filon ? Quels sont les paramètres qui président à l'élaboration de “beaux” cristaux ?
La figure 24 schématise la formation d'un filon dans une roche. Il y a remontée d'un fluide hydrothermal primaire, induit par la géologie régionale (sources, fluides, canaux de transport…). La roche encaissante n'est pas toujours étanche, on peut donc aussi avoir un deuxième flux de fluide issu de la porosité de cette roche. Par ailleurs, le fluide primaire peut réagir chimiquement avec les minéraux de l'encaissant comme nous l'avons vu précédemment.
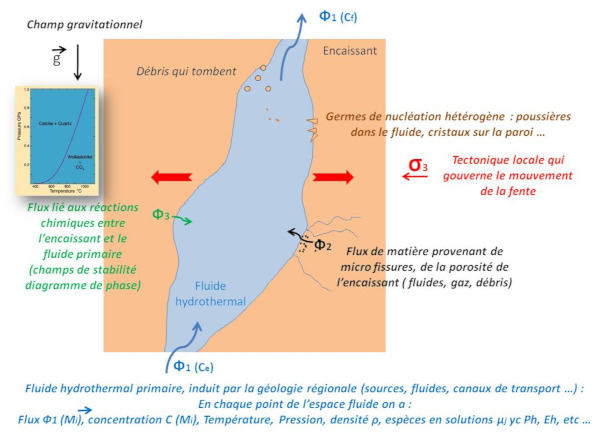
Source - © 2024 — Bernard Barailler
En prenant en compte ces différents flux, en chaque point du fluide, on est face à des conditions particulières de température, pression, espèces en solutions, concentrations, densité, pH, Eh (potentiel d'oxydoréduction)… Ces conditions en chaque point vont conditionner la croissance des faces des cristaux. D'autres facteurs comme le champ de gravité et les dépôts de débris influencent aussi la croissance des cristaux, Ce sont par exemple des dépôts de chlorite (figure 25, quartz avec fantôme de chlorite verte).

Source - © 2009 — Didier Descouens / wikimedia – CC BY-SA 4.0
La partie visible du quartz au premier plan mesure environ 2 cm de “haut”.
L'ensemble est un système dynamique. Le flux peut varier en fonction des conditions sous-jacentes. Les contraintes tectoniques locales sont aussi à prendre en compte. Dans le cas où le flux est constant, c'est-à-dire le nombre d'unités de croissance est constant, si la faille s'écarte plus qu'elle ne se comble par cristallisation de remplissage, alors V augmente et P diminue donc la cristallisation se poursuit. À l'inverse, si la fente se bouche par la cristallisation en cours, l'apparition d'un bouchon aux étages supérieurs, par exemple, V diminue et P augmente (T augmente ou reste constant) et la cristallisation s'arrête ; une redissolution peut même s'amorcer.
La différence initiale de température entre l'encaissant et le fluide induit la cinétique initiale. La capacité de l'encaissant à évacuer la chaleur est ensuite aussi un paramètre important (comme indiqué dans le cas particulier de la boiling zone avec une évacuation facilitée de la chaleur par la phase vapeur).
Par ailleurs, la nature de la gangue (encaissant) influence la nucléation hétérogène. Dans un encaissant granitique, les phénocristaux de quartz du granite sont autant de germes. Les quartz vont se développer à partir de ces nombreux germes présents en surface. On a alors une croissance dite compétitive de nombreux quartz (figure 26).
À contrario, si la gangue est composée par exemple de cristaux de calcite noire chargée d'hydrocarbures et de dolomie, seul le “meilleur” défaut structurel (présentant une énergie minimale G) sert de germe. C'est le cas dans une géode de type septaria (Barailler, 2023(b) [6] – figure 27). La nucléation se fait alors sur ce défaut et le cristal n'est pas en compétition avec d'autres, il concentre les unités de croissance et peut s'exprimer dans toutes les directions.
 Source - © 2024 — Bernard Barailler Cristaux de quelques millimètres dans leurs plus grandes dimensions. |
 Source - © 2022 — D’après Cyril GRANGEON Minerals&Adventures / ebay Dimensions de l’échantillon : 6×4×3,5 cm. |
Conclusion
Les beaux cristaux, généralement “hydrothermaux”, sont souvent le symbole du solide, de l’organisation, de la dureté, du froid, de la stabilité. Pourtant leur formation hydrothermale provient du liquide, du chaos, de la fluidité, de la chaleur… Concernant leur stabilité, la durée de vie d'un cristal peut être définie de façon simplifiée comme le temps durant lequel l'ensemble de ses atomes reste globalement organisé suivant son système cristallin et son habitus au sein de son environnement de création initial.
De façon générale, on peut estimer que les cristaux ont été modifiés au cours des cycles de Wilson liés à l'apparition des supercontinents (Arnould et al., 2024 [4]). En effet, ceux-ci sont métamorphisés et recyclés par les mouvements des plaques tectoniques. De plus, les cristaux ont une durée d'existence “limitée” par les phénomènes de surface que sont l'érosion et l'altération.
Bibliographie
- J.-L. Abbé, J.-F. Berger, P. Blanchemanche, H. Bruneton, L. Chabal, S. Guillon, L. Le Roy, J. Norgari, 2009. L'étang de Montady sur la longue durée, PCR Montady [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- A. Abréal, 2010. Les grenats dans les pegmatites(lien externe - nouvelle fenêtre), J. of Pers. Mineralogist, 13, 149-181 [consulté, novembre 2024]
- G.B. Alexander, W.M. Heston, R.K. Iler, 1954. The Solubility of Amorphous Silica in Water(lien externe - nouvelle fenêtre), The Journal of Physical Chemistry, 58, 6, 453-455 [première page en libre accès]
- M. Arnould, B. Robert, A. Triantafyllou, 2024. Le cycle supercontinental, Planet Terre - ISSN 2552-9250
- B. Barailler, 2023(a). Fleurs d'opale et péridotites du volcan des Baumes, Hérault, Planet Terre - ISSN 2552-9250
- B. Barailler, 2023(b). Les cristallisations rencontrées dans les septarias, une diversité minéralogique à l'échelle (pluri)millimétrique, Planet Terre - ISSN 2552-9250
- B. Barailler, 2024. Pourquoi certains quartz sont-ils “fumés” ?, Planet Terre - ISSN 2552-9250
- R. Bouchet Bert-Manoz, 2014. L'état supercritique en sciences de la Terre, Planet Terre - ISSN 2552-9250
- W.K. Burton, N. Cabrera, F.C. Frank, 1951. The growth of crystals and the equilibrium structure of their surfaces(lien externe - nouvelle fenêtre), Philosophical Transactions of the Royal Society of London. Series A, Mathematical and Physical Sciences, 243, 299-358 [libre accès]
- A.J. Cavosie, J.W. Valley, S.A. Wilde, E.I.M.F., 2005. Magmatic δ18O in 4400–3900 Ma detrital zircons: A record of the alteration and recycling of crust in the Early Archean(lien externe - nouvelle fenêtre), Earth and Planetary Science Letters, 235, 3–4, 663-681
- J. Dan, D.H. Yaalon, 1982. Automorphic saline soils in Israel, in Aridic soils and geomorphic processes, D.H. Yaalon (ed.), Catena, 103-116
- G. Dromart, 2020. La crise de salinité messinienne en Méditerranée, Planet Terre - ISSN 2552-9250
- F. Espitalier, F. Baillon, J. Schwartzentruber, R. David, A. Gaunand, M. Cournil, A. Cameirão, 2024. Les Fondamentaux de la Cristallisation et de la Précipitation(lien externe - nouvelle fenêtre), formation École des Mines d'Albi-Carmaux [consulté, novembre 2024]
- R.O. Fournier, R.W. Potter, 1982. An equation correlating the solubility of quartz in water from 25° to 900°C at pressures up to 10,000 bars(lien externe - nouvelle fenêtre), Geochimica et Cosmochimica Acta, 46, 10, 1969-1973
- R. Kern, 1968. Croissance cristalline et adsorption, Bulletin de la Société française de Minéralogie et de Cristallographie, 91, 217-266 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- M. Kulichenko, N. Fedik, K.V. Bozhenko, A.I. Boldyrev, 2019. Hydrated Sulfate Clusters SO42–(H2O)n (n = 1–40): Charge Distribution Through Solvation Shells and Stabilization(lien externe - nouvelle fenêtre), The Journal of Physical Chemistry B, 123, -4065-4069 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- J.M. Martín, J.C. Braga, C. Betzler, 2002. The Messinian Guadalhorce corridor: the last northern, Atlantic–Mediterranean gateway(lien externe - nouvelle fenêtre), Terra Nova, 13, 418-424
- M. Pelser, J.J. Eksteen, L. Lorenzen, C. Aldrich, 2003. The control of calcium and magnesium in a base metal sulphate leach solution, Proceedings: XXII International Mineral Processing Congress, 1240-1248 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- P.M. Rendel, B.W. Mountain, 2023. Solubility of quartz in supercritical water from 375 °C to 600 °C and 200–270 bar(lien externe - nouvelle fenêtre), The Journal of Supercritical Fluids, 196, 105883
- J.D. Rimstidt, 1997. Quartz solubility at low temperatures(lien externe - nouvelle fenêtre), Geochimica Cosmochimica Acta, 61, 13, 2553-2558
- J.-M. Rouchy, M.-M. Blanc-Valleron, 2006. Les évaporites – Matériaux singuliers, milieux extrêmes, Vuibert, 190p ISBN13: 978-2-7117-5390-1
- D.J. Shearman, 1971. Marine evaporites. The calcium sulphate facies, Unpubl. notebook. The Univ. of Calgary, A.A.P.G. Seminar, 65p
- F. Soleilhavoup, 2011. Microformes d'accumulation et d'ablation sur les surfaces désertiques du Sahara(lien externe - nouvelle fenêtre), Géomorphologie : relief, processus, environnement, 17, 2, 173-186 [libre accès]
- M. Schultz, 2015. Les évaporites de la Vallée de la Mort (Californie), Planet Terre - ISSN 2552-9250
- D. Sibarani, H. Sippola, P. Taskinen, D. Lindberg, 2022. Critical evaluation of CuSO4-H2O system up to solubility limit, from eutectic point to 373.15 K(lien externe - nouvelle fenêtre), Chemical Engineering Science, 257, 117689 [open access]
- P. Thomas, 2005a. Quartz synthétique et quartz naturel, Planet Terre - ISSN 2552-9250
- P. Thomas, 2005b. Champ hydrothermal du Tatio, Andes chiliennes, Planet Terre - ISSN 2552-9250
- P. Thomas, 2005c. Geysérite et eaux siliceuses, Planet Terre - ISSN 2552-9250
- P. Thomas, 2008. Un gisement d'hydrocarbures vu de l'intérieur et un trésor du patrimoine géologique français : la mine de bitume de Dallet (Puy de Dôme), dite « Mine des Rois », Planet Terre - ISSN 2552-9250
- P. Thomas, 2009. Géologie et aménagement du territoire, un exemple de réussite : l'aménagement de l'étang de Montady (Hérault), Planet Terre - ISSN 2552-9250
- P. Thomas, 2015. Empreintes anciennes de halite (pseudomorphoses) en Afrique du Sud et milieux actuels de sédimentation d'évaporites en Égypte et en Grèce, Planet Terre - ISSN 2552-9250
- P. Thomas, 2018. Les cristallisations de halite (NaCl) dans et autour de l'étang de Lavalduc, Bouches du Rhône, Planet Terre - ISSN 2552-9250
- P. Thomas, 2019. Un exemple de salar : le salar d'Atacama, Chili, Planet Terre - ISSN 2552-9250
- P. Thomas, 2021(a). Les filons de calcite associés à la Faille Nord-pyrénéenne, Sournia, Pyrénées-Orientales, Planet Terre - ISSN 2552-9250
- P. Thomas, 2021(b). Filons de quartz des Alpes et d'ailleurs, Planet Terre - ISSN 2552-9250
- J.V. Walther, H.C. Helgeson, 1977. Calculation of the thermodynamic properties of aqueous silica and the solubility of quartz and its polymorphs at high pressures and temperatures(lien externe - nouvelle fenêtre), American Journal of Science, 277, 10, 1315-1351 [libre accès]
- J. Zaccaro, A. Ibanez, 2014. Croissance en solution de cristaux massifs(lien externe - nouvelle fenêtre), L'actualité chimique, 387-388-389, 70-76 [libre accès]
Remerciements
Le texte soumis a bénéficié des commentaires de Benjamin Rondeau (Université de Nantes), Éric Rodari (minéralogiste amateur), Bastien Audran (Géosciences Rennes) et Kim Totaro (Museum national d’Histoire naturelle du Luxembourg).
Cet article a bénéficié d’une relecture approfondie sur le fond et sur la forme par Jean-Philippe Perrillat (LGLTPE, Univ. Lyon 1) et d’une relecture plus formelle mais non moins minutieuse par Cyril Langlois (ENS de Lyon).
[1] L’augmentation de la solubilité lorsque la température augmente est un comportement “classique” mais qui connait quelques exceptions majeures. Ainsi, la solubilité du carbonate de calcium (CaCO3) ou de lithium (LiCO3) diminue, elle, lorsque la température augmente.
[2] Chott ou sebkha : dépression plate généralement proche du littoral, ennoyée par intermittence, au fond recouvert d'une couche de sel. L'alimentation en eau provient selon les cas de la mer, des précipitations, des ruissellements ou de remontées de nappes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
-sulfate-pentahydrate-unit-cell-1985-3D-bs-17.png){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}