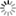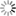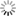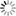Solubilité et solutions aqueuses, rappels et calculs
Article | 02/04/2025
Résumé
Dissolution, cristallisation, constante d'équilibre et solubilité dans un cas simple “XY⟷X+Y”. Illustration rapide avec le gypse.
Table des matières
- Introduction, solubilité du gypse
- Dissolution / cristallisation : équilibre, saturation et nucléation spontanée
- Relation entre les activités des solutés à l'équilibre
- Solubilité et présence de l'un et/ou l'autre des solutés dans la solution initiale
- Calcul des quantités dissoutes ou précipitées, connaissant les teneurs initiales en soluté
- Conclusion et perspectives
Introduction, solubilité du gypse
 Source - © 2016 Pierre Thomas Figure 1. Rigoles de dissolution dans des couches de gypse oligocène du bassin de Mourmoiron (Vaucluse) Le ruissellement des eaux de pluie sur le gypse crée des mini-rigoles de dissolution centimétriques qui se développent en quelques années après que le gypse a été mis à l'affleurement (par un éboulement, des travaux …). Les grandes formations de gypse se retrouvent dans les séries évaporitiques (après les carbonates, avant la halite), comme ici, mais on trouve aussi des cristaux de gypse dans des contextes non-évaporitiques comme dans des marnes lacustres (figure 2) ou formant des filons (figure 3). À retrouver dans Figures de dissolution, de recristallisation et de tectonique dans les gypses de l'Éocène terminal / Oligocène basal de Mormoiron, bassin de Carpentras, Vaucluse. | |
 Source - © 2021 Pierre Thomas |
 Source - © 2013 Pierre Thomas |
Quelle information donne-t-on réellement lorsqu'on dit, par exemple, que le gypse [CaSO4·2H2O] a une solubilité d'environ 2 g/L ?
Tout d'abord, on donne une information incomplète si on ne précise pas qu'on se place dans une solution aqueuse à 20°C à pression “ambiante” (1 atm ou 1 bar). Ces précisions sont importantes en sciences de la Terre puisque l'eau peut se trouver à des températures et pressions très variables, avec de l'eau liquide salée à moins de 0°C jusqu'aux circulations et sources hydrothermales les plus chaudes, et à des pressions de plusieurs atmosphères au fond des océans et dans la croute terrestre. Et encore, on ne parle ici que d'eau liquide “normale” et pas d'eau supercritique, fluide aux capacités de dissolution plus importantes (cf. L'état supercritique en sciences de la Terre.
Même dans les conditions “classiques” et “de surface”, il faut savoir si – comme ce sera le cas dans les parties suivantes – on parle de la solubilité comme un paramètre physique caractéristique (propriété du minéral / solide considéré) ou comme un résultat d'une mesure dans un cas précis (quantité de minéral / solide qu'on arrive à dissoudre dans une solution aqueuse donnée). En effet, dans le premier cas, dans des conditions P et T connues, la solubilité est une constante alors que dans le second elle peut être variable, à P et T connues, en fonction d’autres paramètres de l’expérience, comme nous allons le voir par la suite.
Dissolution / cristallisation : équilibre, saturation et nucléation spontanée
Soit la réaction XYsolide⟷Xaq+YaqXYsolide⟷Xaq+Yaq.
La constante d'équilibre de cette réaction est appelée produit de solubilité, noté KSKS.
On a, par définition, KS=a(Xéq)⋅a(Yéq)/a(XYéq)KS=a(Xéq)⋅a(Yéq)/a(XYéq) [loi d'action de masse], où “aa” désigne l'activité de l'espèce entre parenthèse et “éqéq” l'état d'équilibre.
Pour un corps pur, l'activité est égale à 1, d'où a(XYéq)=1a(XYéq)=1. Pour une solution aqueuse, l'activité de l'eau, composant ultramajoritaire, est égale à 1 et l'activité d'un soluté est alors égale à sa concentration (en mol/L) divisée par une concentration de référence ([C0][C0] (égale à 1 mol/L, le plus souvent) [l'activité n'a pas d'unité].
On a donc a(Xéq)=[Xéq]/[C0]a(Xéq)=[Xéq]/[C0] que l'on notera par simplification XéqXéq par la suite.
On peut alors écrire KS=Xéq⋅YéqKS=Xéq⋅Yéq.
Notons que KSKS est strictement positif, puisqu’une valeur nulle signifierait que le minéral est insoluble. En effet, l’équilibre serait atteint en l’absence de X et/ou de Y en solution (activité nulle), donc en l’absence de dissolution du minéral.
Par ailleurs, on appelle solubilité d'un solide, la quantité de ce solide que l'on peut dissoudre dans une solution, quantité exprimée en g de produit pour 100 g de solvant ou en g par litre de solvant. Remarquons qu'en présence de solide, à l'équilibre, les activités (concentrations) en X et Y sont fixées par le KSKS, la solution est dite saturée (il n’est plus possible de dissoudre davantage de solide). Mais en l'absence de solide (on le retire), si l'on concentre la solution (évaporation partielle, par exemple) alors, thermodynamiquement parlant, il devrait y avoir précipitation de sorte que les concentrations des solutés restent constantes et que le produit des activités soit égal à KSKS. Dans les faits, du fait de barrières énergétiques d'activation, il est possible d'obtenir une solution dite sursaturée dans laquelle l'un et/ou l'autre des solutés est en excès par rapport à l'équilibre, on alors X⋅Y>KSX⋅Y>KS. Une perturbation telle que l'apport d'un germe ou un mouvement brusque dans une telle solution enclenchera alors la cristallisation et une évolution vers une situation d'équilibre. Sans cela, il existe un seuil maximal, appelé seuil de sursaturation ou de nucléation spontanée, seuil auquel la nucléation se produit spontanément. Dans une solution aqueuse ne comportant que les solutés X et Y, on peut ainsi définir le produit KNKN de nucléation spontanée de manière similaire au KSKS, par KN=Xnuc⋅YnucKN=Xnuc⋅Ynuc, XnucXnuc et YnucYnuc étant les activités au seuil de nucléation spontanée. On a, par définition de la (sur)saturation, KN>KSKN>KS.
Relation entre les activités des solutés à l'équilibre
Notons ΣΣ l'activité totale en solutés participant à la réaction, on a Σ=X+YΣ=X+Y.
À l'équilibre, on a un système de 2 équations
- (A) Xéq⋅Yéq=KSXéq⋅Yéq=KS,
- (B) Xéq+Yéq=ΣXéq+Yéq=Σ.
À l'équilibre, les concentrations, et donc les activités, ont des valeurs positives et non nulles (KS>0KS>0).
De (A), on peut donc écrire Yéq=KS/XéqYéq=KS/Xéq.
En injectant cela dans (B), on obtient Σ=Xéq+KS/XéqΣ=Xéq+KS/Xéq.
ΣΣ est une fonction de la variable XéqXéq. Pratiquons une analyse rapide des variations de cette fonction Σ(Xéq)Σ(Xéq) en prenant la notation classique de la forme f(x)f(x), ce qui nous ramène à une petite étude de fonction avec, ici, x>0x>0.
On a f(x)=x+KS/xf(x)=x+KS/x, d'où sa dérivée f′(x)=1−KS/x2 dont on étudie le “signe”.
- f′(x)=0 lorsque 1=KS/x2, soit lorsque que x=√KS.
- Pour x<√KS, on a x2<KS, d'où 1<KS/x2, soit 1−KS/x2<0, on a donc f′(x)<0, la fonction f est alors décroissante.
- Pour x>√KS on montre de même qu’on a f′(x)>0, la fonction f est alors croissante.
- Pour x=√KS, la fonction f prend la valeur f(√KS)=√KS+KS/√KS=2√KS qui est ici une valeur minimale (fonction décroissante puis croissante).
L'activité totale Σ est donc une fonction montrant un minimum lorsque Xéq=√KS, ce minimum étant égal à Σ=2√KS. De l'équation (A) ou (B) on obtient Yéq=√KS. La somme des activités est donc minimale lorsque les solutés sont ici présents en quantités égales.
Solubilité et présence de l'un et/ou l'autre des solutés dans la solution initiale
En partant d'eau pure (“ep”) dans laquelle on dissout du minéral XY, on a à l'équilibre Xéq−ep=Yéq−ep=√KS. La quantité de soluté X et de soluté Y en solution provient totalement de la quantité de minéral XY dissout. On a donc au final une “quantité” Yéq−ep de soluté Y venant entièrement de la dissolution du minéral et égale à √KS.
Partons maintenant d'une solution contenant déjà du soluté X. Lors de la dissolution de XY, des quantités équivalentes des deux solutés sont mises en solution et donc, comme il y a déjà du soluté X au départ mais pas de Y, on aura nécessairement à tout instant X>Y en solution. Ainsi, on arrivera à l'équilibre pour des valeurs X1 et Y1 telles que, à la fois, (α) X1>Y1 et (β) X1⋅Y1=KS. De l'équation (α) on obtient alors X1⋅X1>Y1⋅X1 (en multipliant chaque terme par X1) mais aussi X1⋅Y1>Y1⋅Y1 (en multipliant chaque terme par Y1), d'où, en utilisant l'équation (β), les inégalités X21>KS et KS>Y21, soit encore X1>√KS et Y1<√KS. La “quantité” Y1 de soluté Y en solution vient entièrement de la dissolution du minéral XY et cette quantité est strictement inférieure à √KS, donc inférieure à la quantité libérée en solution en partant d'eau pure. On a donc dissout moins de minéral XY dans de l'eau contenant déjà au moins un peu de soluté qu'en partant d'eau pure. Remarquons que si l'on part d'une solution contenant déjà X et Y en solution, on montrerait de même qu'on obtient une dissolution moindre du fait que le produit final des activités est constant et que les quantités finales de solutés prennent en compte les quantités initialement présentes.
La solubilité d'un minéral est donc la quantité maximale que l'on peut dissoudre dans de l'eau pure. Si la solution initiale contient déjà l'un et/ou l'autre des solutés relâchés par la dissolution, la quantité de minéral qu'il est possible de dissoudre est moindre.
Calcul des quantités dissoutes ou précipitées, connaissant les teneurs initiales en soluté
La connaissance des solubilités des cristaux permet de calculer les caractéristiques finales d'une solution dont on connait les caractéristiques initiales et d'en déduire les quantités dissoutes ou précipitées de minéral.
Soit Xi et Yi, d'une part, et Xf et Yf, d'autre part, les activités initiales et finales des solutés X et Y de la réaction XYsolide⟷Xaq+Yaq dont le produit de solubilité est √KS.
Dans cette réaction simple, les quantités (en moles) de solutés X et Y libérées en solution par dissolution de XY ou retirées de la solution par précipitation sont égales. Leurs variations de concentration en (mol/L) et donc leurs variations d'activité sont donc elles aussi égales. Notons Δ cette différence, Xf−Xi=Yf−Yi=Δ.
Soit Xi et Yi connus, 3 cas se présentent à nous.
- Lorsque Xi⋅Yi=KS, on est à l'équilibre et alors “rien ne se passe”, du moins à l'échelle macroscopique. Toute formation de minéral est immédiatement compensée par la dissolution d'une quantité équivalente de ce même minéral, et inversement. On a alors Xf=Xi et Yf=Yi, et donc Δ=0.
- Lorsque Xi⋅Yi>KS, la solution est sursaturée, il peut y avoir précipitation. On peut calculer les caractéristiques de la solution finale et en déduire la quantité de solutés consommés (Δ<0) et de cristaux qu'il est possible de former avant d'atteindre l'équilibre.
- Lorsque Xi⋅Yi<KS, la solution est sous-saturée, en l'absence de cristaux rien ne se passe, en présence de cristaux il y a dissolution jusqu'à dissolution complète ou jusqu'à équilibre. On peut calculer les caractéristiques de la solution finale et en déduire les quantités de solutés libérés en solution (Δ>0) et la quantité maximale de cristaux qu'il est possible de dissoudre.
La variation Δ permet de remonter à la variation molaire de soluté qui, dans le cas simple étudié, correspond à la variation molaire de minéral, variation à partir de laquelle on peut déterminer la variation massique de minéral (dissout ou cristallisé).
Dans tous les cas, les activités de X et Y évoluent de manière équivalente jusqu'à atteindre l'équilibre. On doit aussi toujours avoir des activités finales strictement positives : les éventuelles solutions mathématiques négatives ou nulles sont donc à exclure. Théoriquement (solution d'eau avec seulement X et Y) le produit des activités initiales ne peut pas dépasser le seuil de nucléation spontanée KN.
On a donc les 3 équations suivantes :
- (1) Xf⋅Yf=KS,
- (2) Xf−Xi=Δ, soit Xf=Xi+Δ,
- (3) Yf−Yi=Δ, soit Yf=Yi+Δ.
En “injectant” (2) et (3) dans (1), on obtient (Xi+Δ)⋅(Yi+Δ)=KS, ce qui, en développant s'écrit encore
- Δ2+(Xi+Yi)⋅Δ+Xi⋅Yi−KS=0.
On obtient une équation du second degré à une inconnue du type
- a⋅x2+b⋅x+c=0.
Les racines (“solutions”) de cette équation sont obtenues en calculant d'abord son déterminant det=b2−4ac. Puis, lorsque det>0, les deux “solutions” mathématiques sont x=(−b±√det)/2a. Si det=0, on a une solution dite double, et si det<0 on a deux solutions “imaginaires” qui ne peuvent indiquer des valeurs “réelles” d'activités et sont donc à exclure.
On a ici, det=(Xi+Yi)2−4⋅(Xi⋅Yi−KS). En développant, on obtient det=Xi2+Yi2+2⋅Xi⋅Yi−4⋅Xi⋅Yi+4⋅KS, soit det=Xi2+Yi2−2⋅Xi⋅Yi+4⋅KS, ce qui peut encore s'écrire
det=(Xi−Yi)2+4⋅KS
ou finalement
det=4(Xi−Yi2)2+4⋅KS=4⋅[(Xi−Yi2)2+KS].
On vérifie alors qu'on a bien det>0 (2 racines non imaginaires).
Les deux solutions mathématiques sont
Δ1=−(Xi+Yi2)−(12)√det, soit Δ1=−(Xi+Yi2)−√(Xi−Yi2)2+KS
et
Δ2=−(Xi+Yi2)+(12)√det, soit Δ2=−(Xi+Yi2)+√(Xi−Yi2)2+KS.
Les activités étant positives et KS strictement positif, on en déduit que la solution Δ1 est toujours strictement négative et ne peut donc pas s’appliquer aux 3 configurations évoquées (on attend une variation négative seulement en cas de solution sursaturée).
Calculons cependant Xf en prenant la solution Δ1. On a alors Xf=Xi+Δ1, soit Xf=Xi−(Xi+Yi2)−√(Xi−Yi2)2+KS, ce qui donne
Xf=(Xi−Yi2)−√(Xi−Yi2)2+KS.
Comme KS>0, on voit que l'activité finale Xfest strictement négative… ce qui exclut définitivement cette solution mathématique Δ1.
Vérifions que la solution Δ2 répond, elle, au 3 cas envisagés.
- Lorsque Xi⋅Yi=KS, on a alors det=(Xi+Yi)2 (d'après la première écriture du déterminant). On a alors Δ2=−(Xi+Yi2)+√(Xi+Yi2)2, soit Δ2=−(Xi+Yi2)+(Xi+Yi2)=0, comme attendu.
- Lorsque Xi⋅Yi>KS, Xi⋅Yi−KS>0, d'où −4⋅(Xi⋅Yi−KS)<0. On a donc (Xi+Yi)2−4⋅(Xi⋅Yi−KS)<(Xi+Yi)2, soit det<(Xi+Yi)2 (première écriture du déterminant) ou √det<(Xi+Yi), ce qui amène, en divisant par 2 chaque terme puis en lui ajoutant −(Xi+Yi2), à −(Xi+Yi2)+(12)√det<0, soitΔ2<0, comme attendu.
- Lorsque Xi⋅Yi<KS, on montre, en procédant comme ci-dessus, qu'alors Δ2>0, comme attendu.
La solution générale unique est donc Δ=−(Xi+Yi2)+√(Xi−Yi2)2+KS.
Ce qui donne les activités finales suivantes :
Xf=Xi−(Xi+Yi2)+√(Xi−Yi2)2+KS et Yf=Yi−(Xi+Yi2)+√(Xi−Yi2)2+KS.
Ce qui au final, peut s'écrire, en remarquant que (Xi−Yi)2=(Yi−Xi)2,
Xf=(Xi−Yi2)+√(Xi−Yi2)2+KS
et
Yf=(Yi−Xi2)+√(Yi−Xi2)2+KS.
On vérifie rapidement que lorsque Xi=Yi, on aboutit à Xf=Yf=√KS avec Δ=−(Xi+Yi2)+√KS.
Lorsque Xi=Yi, on voit que Δ est maximal (et positif) lorsque −(Xi+Yi2) est minimal, soit lorsque Xi=Yi=0, alors Δ=√KS. On retrouve le fait que la dissolution (Δ>0) maximale est obtenue pour une solution initiale d'eau pure lorsque les teneurs initiales en solutés sont égales et nulles.
En général, en dissolution, si Xi≠Yi, en prenant le cas Yi<Xi, les variations d'activités étant égales, on a alors Yf<Xf et donc Yf⋅Yf<Yf⋅Xf, soit Yf<√KS. La variation d'activité Δ=Yf−Yi est inférieure à Yf et donc aussi strictement inférieure à √KS. On retrouve bien que la “quantité” maximale de minéral dissout est égale à √KS et est obtenue par dissolution dans de l'eau pure, mais qu’en présence de l’un et/ou l’autre des solutés dans la solution initiale sous-saturée la “quantité” de minéral dissout est inférieure à √KS.
Conclusion et perspectives
En résumé.
Pour une réaction XYsolide⟷Xaq+Yaq, connaissant le produit de solubilité KS et les activités initiales Xi et Yi, la dissolution de minéral (en présence de minéral à dissoudre) ou la précipitation de minéral (considérée comme effective en cas de sursaturation) aboutissent à un état final décrit par les relations suivantes.
Xf⋅Yf=KS
Xf=(Xi−Yi2)+√(Xi−Yi2)2+KS
Yf=(Yi−Xi2)+√(Yi−Xi2)2+KS
Δ=Xf−Xi=Yf−Yi=−(Xi+Yi2)+√(Xi−Yi2)2+KS
Δ étant la variation d'activité de chaque soluté, qui, exprimée en variation molaire, correspond à la variation de minéral dissout ou précipité.
De plus, on a vu que la somme des activités Xf+Yf=Σ est minimale lorsque Xf=Yf donc aussi lorsque Xi=Yi puisque Xf−Xi=Δ=Yf−Yi.
On a vu que la solubilité d'un minéral, définie en lien avec le produit de solubilité, correspond à la quantité maximale qu'il est possible de dissoudre dans de l'eau pure. Pour une solution initiale contenant déjà l'un des solutés issus de la dissolution de ce minéral, la solubilité “apparente” (quantité de minéral qu'on va pouvoir dissoudre) est inférieure à la solubilité ainsi définie.
On pourrait calculer la quantité de minéral XY précipité lors du passage du seuil de nucléation spontanée à équilibre. Il suffirait d'ajouter la condition initiale entre les activités correspondant à cette situation, à savoir Xi⋅Yi=KN.
Dans les calculs, on a considéré la dissolution comme possible pour déterminer la quantité maximale soluble. On peut calculer les activités finales en cas de “déficit” de minéral (le Δ calculé correspond alors à une quantité supérieure à celle disponible). On détermine alors le Δ “vrai” à partir de la quantité de minéral disponible et on en déduit les activités finales.
Dissolution et cristallisation peuvent se produire dans des solutions dans lesquelles les solutés ne sont pas présents en quantités relatives correspondant aux rapports stœchiométriques de la réaction (ici 1:1 dans le cas simple étudié). Ainsi, par exemple, le minéral XY peut précipiter à partir d'une solution beaucoup plus riche au départ en X qu'en Y… mais alors il ne précipitera qu'une “petite” quantité de minéral avant d'atteindre la saturation.
On a montré ici qu'il est possible de quantifier les évolutions de solutions en contact avec des minéraux solubles ou en capacité de précipiter des minéraux. Pour cela on a pris un cas mathématiquement simple. Les calculs deviennent cependant rapidement plus compliqués dès lors que la réaction est plus “complexe” : coefficients stœchiométriques différents, solutés comme produits et comme réactifs…
Revenons au gypse et à sa solubilité de 2 g/L évoquée en introduction. Cette donnée caractéristique permet de connaitre la quantité molaire de gypse soluble dans de l'eau pure (dans les conditions “de surface”).
La réaction à prendre en compte est CaSO4⋅2H2Osolide⟷Ca2+aq+SO2−4aq+2H2O. Les activités du gypse solide et de l'eau étant égales à 1, on retrouve, comme dans l'exemple traité KS=Ca2+⋅SO42−. Avec une masse molaire d'environ 172 g/mol, une solubilité de 2 g/L indique que l'eau pure peut contenir 2/172 = 11,63.10−3 mol/L de gypse dissout. À l'équilibre, la solution contiendra donc 11,63.10−3 mol/L de Ca2+ et autant de SO42−, on aura donc des activités égales 11,63.10−3 pour ces deux solutés, d'où un produit de solubilité KS égal à 11,63.10−3× 11,63.10−3 soit KS=1,35.10−4. La solution la moins concentrée à l'équilibre avec le gypse solide, ne contenant que les solutés Ca2+ et SO42−, a une concentration totale de 23,26.10−3 mol/L. De plus, on sait que si on a une solution initiale sous-saturée contenant déjà Ca2+ et/ou SO42− alors on ne pourra y dissoudre qu'une quantité de gypse inférieure à 2 grammes par litre. D'autre part, une solution initiale très riche en Ca2+, par exemple, sera “rapidement” saturée, voire sursaturée, avec un faible apport de SO42−.
Notons ici que pour des solutions “réelles”, la présence d'autres solutés non impliqués dans la réaction peut agir comme un inhibiteur de nucléation. Par exemple, la présence de Mg2+ en solution “retarde” la précipitation de gypse, le seuil de saturation et même de nucléation spontanée pouvant être dépassés dans cette solution. Ainsi le gypse est (sur)saturé dans l'eau de mer mais sa précipitation dans la série évaporitique n'a lieu qu'après que les carbonates, dont la dolomite [CaMg(CO3)2] ”pompant” le magnésium, ont précipité.