Article | 29/09/2022
La Fontaine de Réotier (Hautes-Alpes)… et l'acidification des océans
29/09/2022
Résumé
Un exemple local pour comprendre pas à pas le système carbonate avant de changer d’échelle en s’intéressant à l’acidification des océans en lien avec les effets des rejets de CO2 anthropiques.
Table des matières

Source - © 2022 Gil Michard
Voir aussi la géologie et le contexte régional en relisant La fontaine pétrifiante de Réotier, Hautes-Alpes ».
Comment l'eau de la fontaine qui ne dépose rien au niveau de la source, située quelque mètres un peu plus haut, peut-elle donner naissance à ces gigantesques concrétions quelques mètres en contrebas ? Pour l'expliquer, le chimiste va faire intervenir un ensemble de réactions qualifiées de « système carbonate » ; ces mêmes réactions vont gouverner des problèmes à l'échelle mondiale comme, par exemple, l'acidification des océans.
À la source de la Fontaine de Réotier
Tableau 1. Analyse de l'eau de la source de Réotier
mg/L | mmol/L | mmol/L | ||
Sodium | Na+ | 662 | 28,8 | 0,22 |
Potassium | K+ | 17,5 | 0,45 | 0,03 |
Calcium | Ca2+ | 580 | 14,5 | 0,33 |
Magnésium | Mg2+ | 54 | 2,25 | 0,14 |
Chlorure | Cl− | 983 | 27,7 | 0,16 |
Sulfate | SO42− | 1286 | 13,4 | 0,09 |
Hydrogénocarbonate | HCO3− | 506 | 8,3 | 0,86 |
La valeur moyenne des eaux continentales figure en bleu (colonne de droite).
Autres mesures : pH = 6,40 et température = 20°C.
L'eau de la source de Réotier a une composition très particulière. Elle pourrait être considérée comme une eau minérale. La conversion des données classiques en mg/L en unités plus adaptées (mmol/L) présente des avantages :
- vérifier que la somme des charges positives des cations (ici 62,75 milliéquivalent par litre[1] est égale à la somme des charges négatives des anions (62,70 meq/L). En effet, les lois de l'électrostatique imposent que la solution soit électriquement neutre ;
- montrer que les concentrations en Cl− et Na+, d'une part, et en SO42− et Ca2+, d'autre part, sont très voisines. Cela suggère un lessivage en profondeur par l'eau d'évaporites riches en gypse (CaSO4 hydraté) et sel (NaCl), évaporites très présentes localement.
La donnée “hydrogénocarbonate” (encore parfois appelé bicarbonate) associée au pH (acide) indique (voir plus loin) que la quantité de CO2 sous forme de gaz dissout est de 4,9 mmol/L. Cette concentration correspondrait à un équilibre avec une atmosphère contenant 14,5 % de CO2, donc très différente de celle de l'air (0,04 %).
Le système carbonate
Les réactions qui interviennent à Réotier concernent le « système carbonate » : le dioxyde de carbone CO2 est présent dans l'air, dans l'eau (sous forme de CO2 dissout, d'ion hydrogénocarbonate HCO3− et d'ion carbonate CO32−) et dans le solide (CaCO3 et C organique).
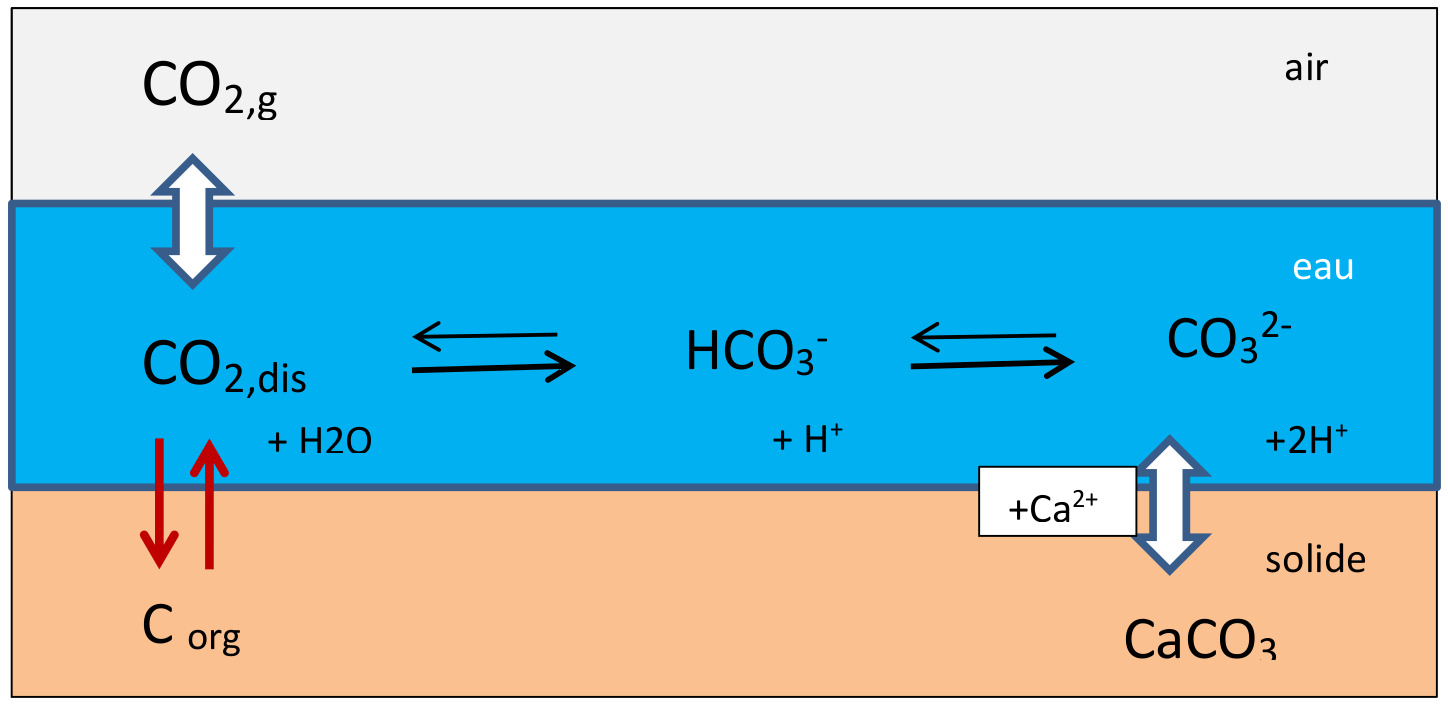
Source - © 2022 Gil Michard
Figure 2. Schéma du système carbonate
Dans les eaux, l'ion hydrogénocarbonate HCO3− est généralement l'espèce prédominante, le CO2 dissout noté CO2,dis peut être échangé avec l'air, consommé par la photosynthèse ou produit par la respiration, CO32− peut réagir avec l'ion calcium et former du carbonate de calcium CaCO3 solide parce qu’insoluble.
Les 3 espèces dissoutes sont en équilibre, équilibre atteint instantanément : CO2,dis + CO32− + H2O ↔ 2 HCO3−, et leurs concentrations sont reliées par la constante d'équilibre K selon la relation suivante :
(équation 1).
Les 3 espèces dissoutes (CO2,dis, HCO3− et CO32−) ne peuvent être dosées directement.
Les mesures possibles du système carbonate sont celles de la concentration totale en carbonate (ΣCO2), de l'alcalinité et du pH.
Il est sans doute nécessaire de bien définir ces trois quantités mesurables.
La concentration totale en carbonate (ΣCO2)
Quand la solution échange du CO2 avec l'air ou le solide, c'est la quantité totale de CO2 par litre d'eau, c'est-à-dire la somme des concentrations des 3 espèces dissoutes, somme notée ΣCO2, qui rend compte quantitativement de cet échange.
(équation 2)
On peut la mesurer en acidifiant la solution puis en chassant les gaz pour finalement mesurer par absorption dans l’infra-rouge le CO2 total libéré.
L'alcalinité
Comme la majorité des eaux naturelles, l'eau de Réotier (Tableau 1) contient essentiellement 7 éléments, dont 6 : le sodium, le chlorure, le calcium, le sulfate, le magnésium et le potassium sont toujours présents sous la même forme ionique, respectivement Na+, Cl−, Ca2+, SO42−, Mg2+ et K+. Pour une composition donnée en ces éléments, l'ensemble de ces ions portera donc une charge (positive à Réotier, comme c'est généralement le cas). Le dernier élément peut se trouver sous forme de CO2,dis, de HCO3− ou de CO32−. Les proportions de ces 3 espèces s'ajusteront de façon à ce que la solution soit électriquement neutre, c'est-à-dire que la somme des charges négatives des espèces du carbone compense la charge positive de l'ensemble des autres ions. On pose l'équation suivante :
(équation 3)
“ALC” est appelée l'alcalinité de la solution.
L'égalité avec le terme de gauche montre que l'alcalinité ne dépend que de la concentration des espèces dissoutes autres que celle du système carbonate.
L'égalité avec le terme de droite est une équation contribuant à la détermination des concentrations en hydrogénocarbonate et carbonate.
Pour une concentration totale en CO2 (ΣCO2) et une alcalinité (ALC) données, les 3 équations (1), (2) et (3) permettent de calculer les concentrations des 3 espèces. On mesure l'alcalinité par un titrage par une solution d'acide, suivi au pH-mètre.
La meilleure solution pour étudier le système carbonate d'une eau serait de mesurer ALC et ΣCO2, mais cette dernière mesure, bien que réalisable, nécessite d'éviter tout échange de CO2 avec l'air depuis le prélèvement jusqu'à la fin de la mesure.
Le pH
Supposons que l'on ait mesuré ΣCO2 et ALC. Les concentrations [CO2,dis], [HCO3−] et [CO32−] sont alors connues, on peut calculer [H+] par l'une des relations suivantes :
(équation 4)
(équation 5)
K1 et K2 étant des “constantes” dont la valeur en fonction de la température est connue. Elles dépendent aussi, pour les solutions concentrées, de la salinité de la solution. Elles peuvent également être déterminées expérimentalement au cours de la mesure de l'alcalinité.
Comme la détermination des 3 concentrations utilise la relation (1), si on calcule [H+] par la relation (5), la relation (4) est automatiquement vérifiée et vice-versa.
Le pH (égal à −log[H+]) peut être calculé et l'ensemble des 2 derniers paragraphes montre bien que le pH est une conséquence de la composition de la solution.
Mais inversement, à l'aide de ces relations, la mesure du pH (pH-mètre, voire papier pH moins précis mais suffisant pour déceler des variations notables de pH), associée à la température, permet de connaitre les rapports [HCO3−] /[CO2,dis] et [CO32−]/[HCO3−], et la mesure de l'alcalinité permet de connaitre la somme [HCO3−] + 2 [CO32−].
On peut donc, en mesurant pH et alcalinité, obtenir les concentrations des 3 espèces, ainsi que leur somme ΣCO2.
À Réotier, ce sont pH et alcalinité qui ont été mesurés.
Prélèvements et évolution de la composition de l’eau, de la source au bassin inférieur de la Fontaine de Réotier
Points de prélèvements et mesures

Source - © 2022 Gil Michard
Figure 3. Localisation des prélèvements sur le site de la Fontaine de Réotier
Le pH, l'alcalinité et la température de l'eau ont été mesurés en 5 points : à la source (A), sur la gargouille (B), au pied de la chute (C), en dessous de la vasque (D) et dans le bassin inférieur (E).
Le pH, l'alcalinité et la température de l'eau ont été mesurés en 5 points : à la source (A) une quinzaine de mètres en amont de l’extrémité de la gargouille, à l’extrémité de la gargouille (B), au pied de la chute (C), en dessous de la vasque (D) et dans le bassin inférieur (E). Tous les échantillons ont la même teneur en chlorure – teneur anormalement élevée, ce qui montre que l'eau ne subit pas de mélange lors de son parcours. Les variations de composition sont dues à des réactions internes à l'eau, et à des échanges avec l'air et le solide.
Tableau 2. Mesures effectuées aux points de prélèvement de la Fontaine de Réotier
Points prélevés | A | B | C | D | E |
température (°C) | 20,5 | 20,5 | 20,5 | 20 | 19 |
pH | 6,4 | 6,95 | 7,9 | 7,9 | 7,9 |
alcalinité (mmol/L) | 8,15 | 8,05 | 7,02 | 6,25 | 6,07 |
Ces données permettent de calculer les concentrations des 3 espèces carbonatées, et d'en déduire la quantité totale de CO2 dissout (équation 2).
On peut aussi déterminer la pression de CO2 [p(CO2)eau] dans l'eau, calculée à partir de la concentration de CO2,dis et de la température.
Tableau 3. Grandeurs déterminées par le calcul à partir des données du Tableau 2
A | B | C | D | E | ||
HCO3− | mmol/L | 8,13 | 7,98 | 6,49 | 5,78 | 5,61 |
CO2,dis | mmol/L | 4,9 | 1,35 | 0,12 | 0,11 | 0,11 |
CO32− | mmol/L | 0,015 | 0,04 | 0,26 | 0,23 | 0,22 |
ΣCO2 | mmol/L | 13,04 | 9,37 | 6,88 | 6,12 | 5,94 |
p(CO2)eau | hPa | 145 | 40 | 3,7 | 3,3 | 3,2 |
Les 3 dernières lignes, CO32−, ΣCO2 et p(CO2)eau, sont reprises par la suite pour comprendre l'évolution de la composition.
Pertes de CO2 total par l'eau
Du tableau précédent, on reprend la ligne concernant la quantité totale de CO2 dissout (ΣCO2) par litre d'eau. L'eau perd du CO2 total le long de son parcours ; cette perte se fait au bénéfice soit de l'air (dégazage), soit du solide (calcaire – précipitation – ou matière organique – photosynthèse).
Tableau 4. Calcul des pertes de la quantité totale de CO2 dissout
Entre | A et B | B et C | C et D | D et E | |
pertes totales ΣCO2 | mmol/L | 3,67 | 2,49 | 0,75 | 0,18 |
Les pertes sont surtout notables en amont de la chute (point C).
Les équilibres chimiques en solution permettent d'étudier les possibilités de dégazage et de précipitation de carbonate de calcium.
Possibilités de dégazage
Pour voir si l'eau peut perdre du CO2 par dégazage au contact de l'air, il faut comparer les pressions partielles de ce gaz dans l'eau et dans l'air. L'atmosphère étant rapidement mélangée, la pression partielle dans l'air est voisine de 0,4 hPa (concentration de 415 ppmv et pression totale à l'altitude Réotier de 900 hPa).
La pression partielle de CO2 dans l'eau est calculable si l'on connait la concentration de CO2,dis et la température ; les résultats du calcul en chaque point figurent dans le tableau 3 et sont repris ci-dessous.
Tableau 5. Pression partielle de CO2 dans l'eau
A | B | C | D | E | ||
p(CO2)eau | hPa | 145 | 40 | 3,7 | 3,3 | 3,2 |
Le dégazage est possible si la pression partielle de CO2 dans l'eau est supérieure à la pression partielle de CO2 dans l'air. Dans le cas contraire, il y a dissolution du gaz dans l'eau. Le dégazage sera favorisé quand l'eau sera divisée en gouttelettes (augmentation de la surface d’échange eau-air) comme au niveau de la chute (point C).
L'eau de Réotier est très nettement sursaturée, surtout à la source (point A), et dégazera certainement[2]. La baisse de la pression partielle de CO2 au cours du parcours de l'eau semble le confirmer.
Toutefois, des végétaux et des bactéries peuvent utiliser une eau aussi riche en CO2 pour effectuer la photosynthèse. Dégazage et photosynthèse ayant le même effet sur la chimie de l'eau, la distinction ne se fera qu'en fin de la discussion.
Possibilités de précipitation de CaCO3
La forte diminution de CO2,dis le long du parcours de l'eau entraine un déplacement vers la gauche de l'équilibre
CO2,dis + CO32− + H2O↔ 2 HCO3−
, entrainant une légère augmentation de CO2,dis, qui ne compense pas la perte par dégazage, et une augmentation de la concentration en CO32−.
Le Tableau 6 extrait du Tableau 3 confirme l'augmentation de [CO32−].
Tableau 6. Concentration de l'ion carbonate
A | B | C | D | E | ||
CO32− | mmol/L | 0,015 | 0,04 | 0,26 | 0,23 | 0,22 |
La calcite peut précipiter si [CO32−] est supérieure à une concentration limite [CO32−]sat qui dépend de la température, de la concentration en calcium, mais aussi de la salinité. La concentration limite [CO32−]sat déterminée par le calcul est entachée d'une incertitude importante.
Une expérience, dite de « saturométrie », permet de montrer que l'eau à l'émergence a une concentration en carbonate proche de la valeur limite. On mesure le pH de l'eau avant et après addition d'un peu de calcite solide :
- si la solution était sous-saturée, la calcite se dissoudrait et entrainerait une augmentation du pH ;
- si la solution était sursaturée, l'addition de calcite favoriserait sa précipitation et entrainerait une diminution du pH.
L'expérience, faite avec un indicateur coloré, ne montre pas de modification sensible du pH et on peut considérer que l'eau de la source du Réotier est au voisinage de l'équilibre avec la calcite et que la valeur de [CO32−] au point A est la concentration limite [CO32−]sat. Dès le point B, et surtout après le dégazage important qui se produit au pont C, [CO32−] est donc nettement supérieure à [CO32−]sat. On prévoit donc que la calcite peut précipiter ce qui est en accord avec les observations.
Estimation de la quantité de CaCO3 précipitée
Reprenons l'expression de l'alcalinité (équation 3).
En reprenant les résultats des analyses suivons l'évolution de l'alcalinité (Tableau 7 extrait du Tableau 2).
Tableau 7. Évolution de l'alcalinité
A | B | C | D | E | ||
alcalinité | mmol/L | 8,15 | 8,05 | 7,02 | 6,25 | 6,07 |
La diminution observée de l'alcalinité peut être attribuée à la précipitation de CaCO3 car l'analyse du dépôt solide montre qu'il est constitué de CaCO3 pur. Cette estimation par l'alcalinité est plus précise que la détermination directe par la teneur en calcium.
Une précipitation d'1 mmol de CaCO3 par litre entrainera une diminution de la concentration de Ca d'1 mmol/L et la diminution de l'alcalinité de 2 mmol/L.
Tableau 8. Tableau 8. Évaluation des dépôts de CaCO3
Entre | A et B | B et C | C et D | D et E | |
dépôt CaCO3 | mmol/L | 0,05 | 0,51 | 0,38 | 0,09 |
dépôt CaCO3 | kg/jour | 0,1 | 1,5 | 1,1 | 0,3 |
On peut alors calculer les quantités de CaCO3 déposées entre 2 points de prélèvement et compte tenu du débit mesuré à 20 L/min, les quantités déposées par unité de temps.
Estimation de la quantité de CO2 dégazée
Connaissant les pertes de CO2 totales, et celles dues à la précipitation de CaCO3, on obtient par différence la quantité de CO2 dégazée (on y inclut les éventuels effets de la photosynthèse).
Tableau 9. Bilan des réactions
Entre | A et B | B et C | C et D | D et E | |
pertes totales ΣCO2 | mmol/L | 3,67 | 2,49 | 0,75 | 0,18 |
dépôt CaCO3 | mmol/L | 0,05 | 0,51 | 0,38 | 0,09 |
dégazage | mmol/L | 3,62 | 1,98 | 0,37 | 0,09 |
dégazage / dépôt | 72,4 | 3,8 | 1 | 1 | |
dégazage | kg CO2/jour | 4,6 | 2,5 | 0,4 | 0,1 |
Sur le dos de la gargouille (en amont de C), la perte de CO2 est essentiellement due au dégazage. Après la chute (C), dégazage et précipitation se produisent à la même vitesse, ce qui correspond au schéma réactionnel :
2 HCO3− + Ca2+ → CO2,gaz + CaCO3 +H2O.
Bilan des réactions d'échange
Tableau 10. Estimation en masse des quantités de CO2 échangées
Entre | A et B | B et C | C et D | D et E | |
dépôt CaCO3 | kg CaCO3/jour | 0,1 | 1,5 | 1,1 | 0,3 |
dégazage | kg CO2/jour | 4,6 | 2,5 | 0,4 | 0,1 |
photosynthèse | kg végétal/jour | 40 | 25 | 4 | 1 |
Photosynthèse et dégazage ont un effet analogue sur la chimie de la solution. Le bilan ci-dessus permet de préciser la nature de la perte de CO2. Par exemple, si la perte de CO2 entre A et B était due à la photosynthèse il faudrait produire environ 3 kg de matière sèche. Sachant que la matière sèche représente environ 7 % de la matière végétale, il devrait donc se former de l'ordre de 40 kg de matière végétale par jour. Cette estimation n'est strictement valable que pour le jour de l'analyse, mais des analyses qualitatives faites à diverses reprises montrent qu'il n'y a pas de variations très notables depuis 50 ans. Il est donc très improbable que la photosynthèse joue un rôle significatif dans la perte de CO2. Par contre un dégazage total de près de 8 kg de CO2/jour dans l'atmosphère, ce qui correspond aux émissions d'une voiture moyenne parcourant 50 km, ne va guère modifier l'atmosphère et est tout à fait envisageable.
Il est plus difficile de se rendre compte si la quantité de calcite effectivement précipitée est en accord avec la quantité calculée.
En résumé, pour la source du Réotier
La source du Réotier est extrêmement chargée en CO2 dissout. Au contact de l'air, ce CO2 va partir dans l'air, ce qui augmente en retour la concentration d'ion carbonate (CO32−). Celle-ci va dépasser le seuil entrainant le dépôt du carbonate de calcium (CaCO3) qui forme les concrétions caractéristiques de la fontaine. Le site de Réotier est un cas où l'évolution du système carbonate est quantitativement très importante.
On en tire des leçons pour comprendre le comportement de ce système dans les eaux naturelles.
- Deux mesures en plus de la température (ici pH et alcalinité) sont nécessaires et suffisantes pour calculer toutes les autres grandeurs du système carbonate.
- L'évolution des concentrations totales de CO2 dissout (ΣCO2) et de l'alcalinité de la solution permet de quantifier les transferts entre l'eau, l'air et le solide.
- La comparaison de p(CO2)eau et de p(CO2)air, d'une part, et de la concentration de CO32− avec la concentration à saturation, d'autre part, permet de prévoir le sens des réactions de dégazage/dissolution de CO2 et de précipitation/ dissolution de CaCO3.
- Les autres grandeurs comme le pH sont des grandeurs dérivées qui ne sont utiles que parce qu'elles peuvent être mesurées et permettent de déterminer complètement l'état du système carbonate.
Ces leçons vont permettre une toute première approche de l'acidification de l'océan.
Cycle du CO2 et acidification de l'océan
Cycle du CO2 dans l'océan préindustriel
Le Tableau 11 présente des données moyennes estimées de ΣCO2 et d'alcalinité dans l'eau de surface et dans l'eau de fond des océans à l'époque préindustrielle. Y figure aussi la teneur en 14C, qui permet d'estimer les quantités de C échangées par an entre les 2 réservoirs.
Tableau 11. Données caractéristiques de l'océan de surface et de l'océan profond, à l'époque préindustrielle
temp. (°C) | ΣCO2 (mol/m3) | ALC (eq/m3) | R14C | |
surface | 18 | 2 | 2,3 | 0,95 |
fond | 2 | 2,3 | 2,4 | 0,8 |
R14C est le rapport de la teneur en 14C de l'échantillon à celle de l'air.
Partons de l'océan profond : l'eau qui remonte dans la zone superficielle perd de l'alcalinité du fait d'une précipitation de CaCO3, et perd du CO2 total du fait d'une précipitation de CaCO3 et d'une formation nette (non compensée par une minéralisation dans la zone de surface) de matière organique par photosynthèse.
On se retrouve dans une situation similaire à celle de Réotier avec une diminution de la quantité totale de CO2 dissout (la photosynthèse joue le rôle principal dans le cas de l'océan) qui entraine la précipitation de CaCO3 (réalisée ici principalement par des organismes vivants microscopiques : foraminifères, coccolithophoridés…) qui se traduit par une diminution de l'alcalinité.
Le débit du ruisseau est remplacé par le volume d'eau échangé annuellement entre surface et fond qui est estimé par le 14C à 1,5.1015 m3/a. Les quantités précipitées dans la zone de surface tombent par gravité dans le fond où elles se dissolvent. Les quantités de C échangées (en 1015 mol/a) sont portées sur la figure 4.
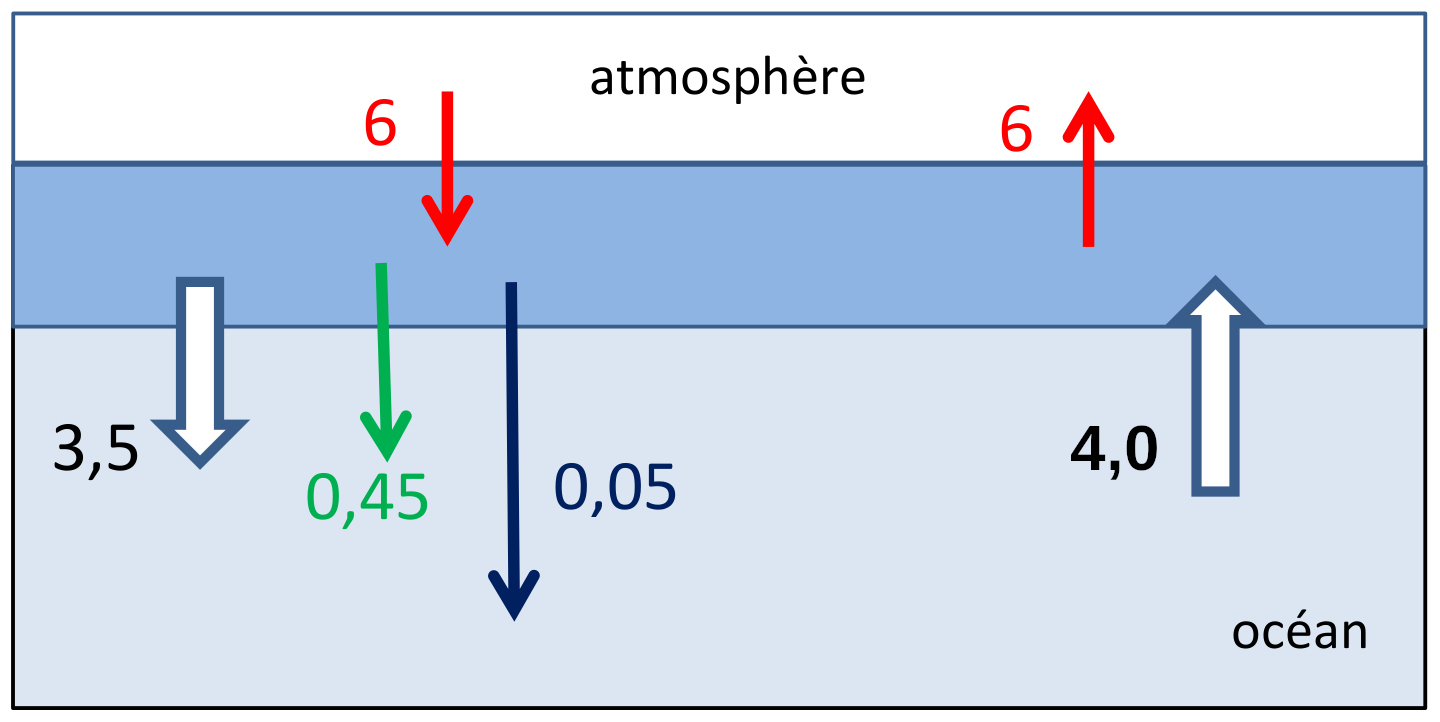
Source - © 2022 Gil Michard
Figure 4. Schéma simplifié du cycle du carbone dans l'océan
Les quantités échangées sont en 1015 mol/a.
Sont représentés les réservoirs “atmosphère”, “océan superficiel” et “océan profond”.
Flèches rouges = échanges gazeux, flèches blanches = échanges de substances dissoutes, flèche verte = chute de matière organique, flèche noire = chute de calcaire (solide).
La teneur en CO2 dans l'atmosphère à l'équilibre avec l'eau de mer de surface, était de 280 ppmv. Comme la quantité totale de CO2 dans l'atmosphère est 30 à 40 fois inférieure à celle de l'océan, la teneur dans l'atmosphère était une conséquence du cycle océanique du CO2.
L'acidification actuelle de l'océan
On brule chaque année 0,7.1015 moles de carbone fossile, donc des quantités du même ordre de grandeur que les échanges naturels. Cela fait croitre la teneur en CO2 de l'atmosphère et une partie de ce CO2 se dissout dans l'océan.
Les mesures des teneurs en CO2 de l'atmosphère sont effectuées en routine en plusieurs points du globe. Les mesures sur le site de Mauna Loa à Hawaï ont débuté en 1958. L'atmosphère se mélange rapidement et l'évolution des valeurs est la même en tous les points de mesure du globe.
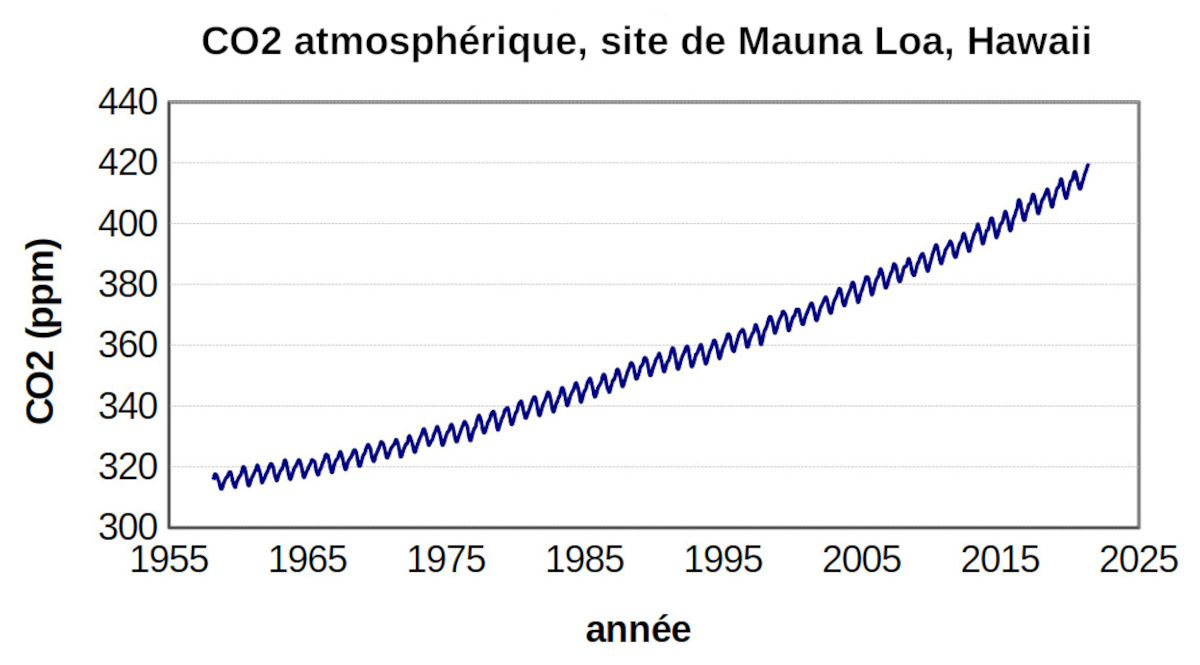
Figure 5. Évolution de la teneur en CO2 de l'atmosphère à Mauna Loa (Hawaï) de 1958 à 2022
Les évolutions annuelles sont représentatives de celle de l'ensemble de l'atmosphère terrestre.
Les estimations globales dans l'océan sont difficiles car les valeurs dépendent du temps et de l'espace. Des mesures mensuelles de ΣCO2 et de l'alcalinité sont effectuées en quelques points de l'océan, au centre de gyres où les courants sont minimum, comme à la station BATS (Bermuda Atlantic Time-series Study). Des évolutions sont clairement perceptibles et correspondent à des évolutions à l'échelle globale.
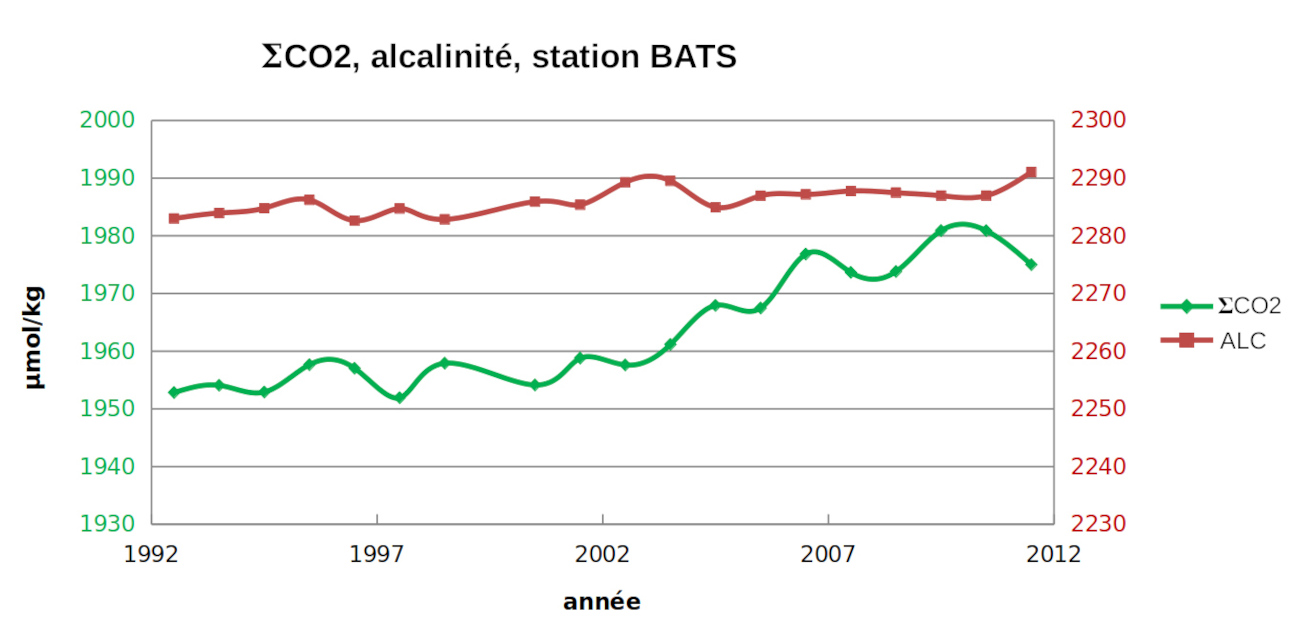
On peut calculer l'évolution des concentrations des espèces dissoutes.
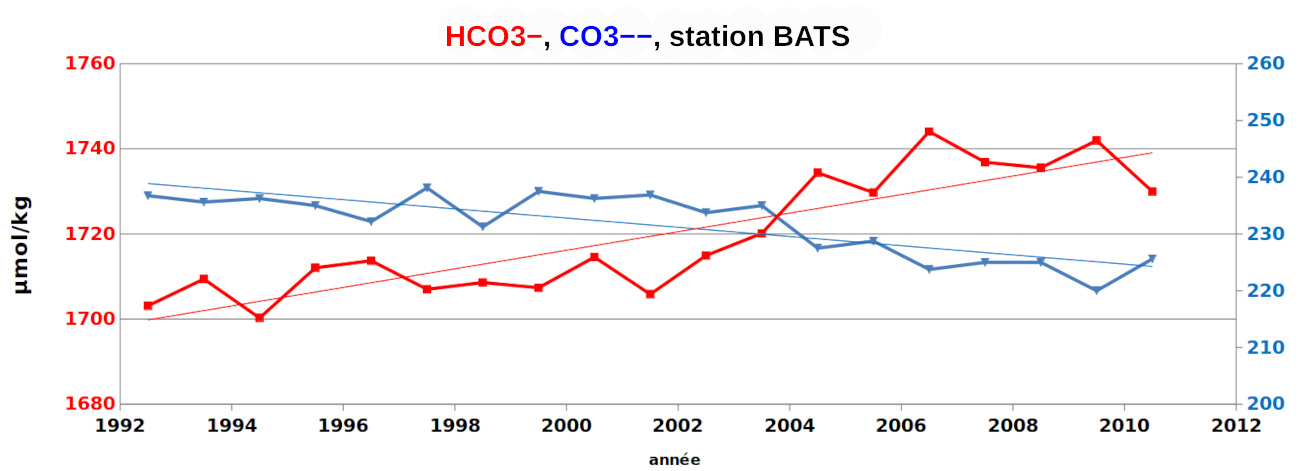
Comme prévisible, l'addition de CO2 entraine un déplacement de la réaction d'équilibre vers la droite :
CO2,dis + CO32− + H2O → 2 HCO3−,
ce qui augmente la concentration de HCO3− et diminue celle de CO32−. Il en résulte une diminution du pH (pH = cste+ log([CO32−]/[HCO3−]).
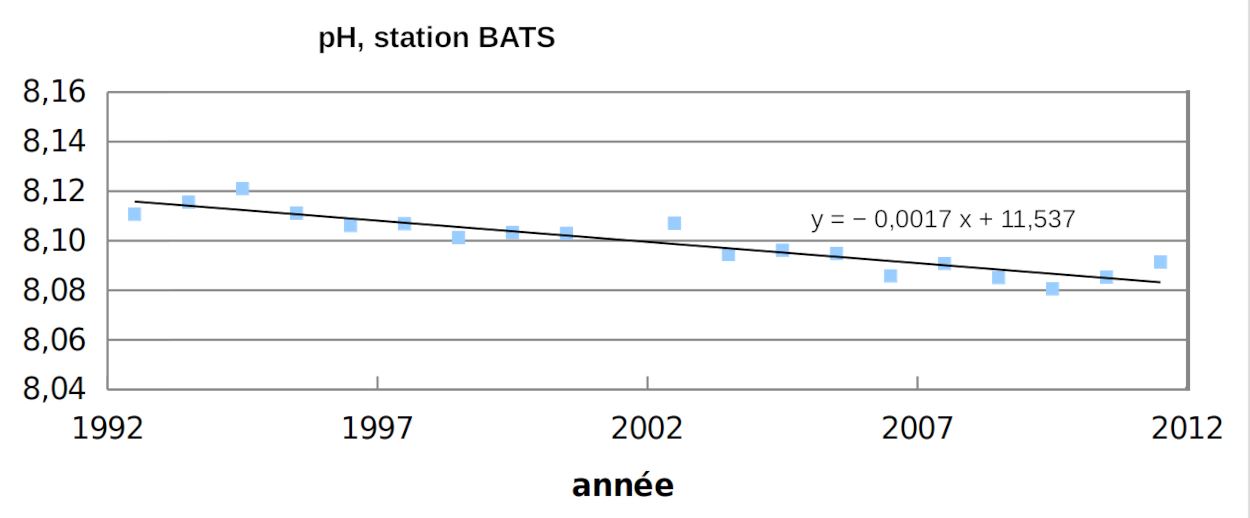
On peut confirmer que les évolutions observées à la station BATS (au large des Bermudes) sont bien représentatives d’une évolution globale en regardant les résultats de l’autre grande station de mesure océanique, la station ALOHA (au large d’Honolulu).

Source - © 2021 PMEL-NOAA
On retrouve bien la tendance à l’enrichissement en CO2 de l’océan (qui suit celui de l’atmosphère) et la baisse du pH de l’océan (acidification).
Source - © 2021 PMEL-NOAA
Localisation par fichier kmz des stations océaniques BATS (Bermudes) et ALOHA (Hawaï).
On estime qu'à l'heure actuelle, l'océan global absorbe entre 25 et 30 % du CO2 anthropique. Les prévisions de l'évolution future nécessitent des évaluations de l'évolution de la température, de la photosynthèse, de la vitesse de formation des coquilles calcaires, de la circulation des océans et surtout la consommation anthropique de carbone fossile.
La chimie donne au moins une certitude : la diminution de la concentration de CO32− va diminuer la proportion de CO2 atmosphérique absorbée par l'océan, océan qui deviendra donc de moins en moins un puits de carbone… Ce qui ne va pas arranger le réchauffement climatique si on continue à rejeter du CO2.
[1] Pour les ions, il peut être judicieux de connaitre la charge induite par un ion dans une solution afin de vérifier la neutralité électrique de cette dernière. Ainsi un “équivalent” est une “mole de charge”. Par exemple, une concentration de 20 mmol/L de Ca2+ correspond à 40 meq/L puisque chaque ion porte 2 charges. Il faudra donc 40 meq/L d'anions pour neutraliser cette charge.
[2] Si p(CO2)eau était supérieure à la pression atmosphérique (900 hPa à l'altitude de Réotier), le CO2 sortirait sous forme de bulles. Là, le dégazage se produit par des échanges de gaz à l'interface entre l'eau et l'air.