
Article | 04/06/2025
Un cristal monoclinique exemplaire – Comment se forment les gypses du Trièves
04/06/2025
Auteur(s) / Autrice(s) :
Publié par :
- Olivier DequinceyENS de Lyon / DGESCO
Résumé
Un modèle de cristallisation du gypse basé sur l’osmose inverse. Étude des gypses monocliniques au sein des varves de Sinard (Isère).
Introduction

Source - © 2024 — Bernard Barailler
Ce monocristal mesure environ 7×3 cm.
Il est rare de trouver un cristal dont la forme est exactement celle de son système cristallin. La figure précédente montre un cristal de gypse avec une forme monoclinique exemplaire.
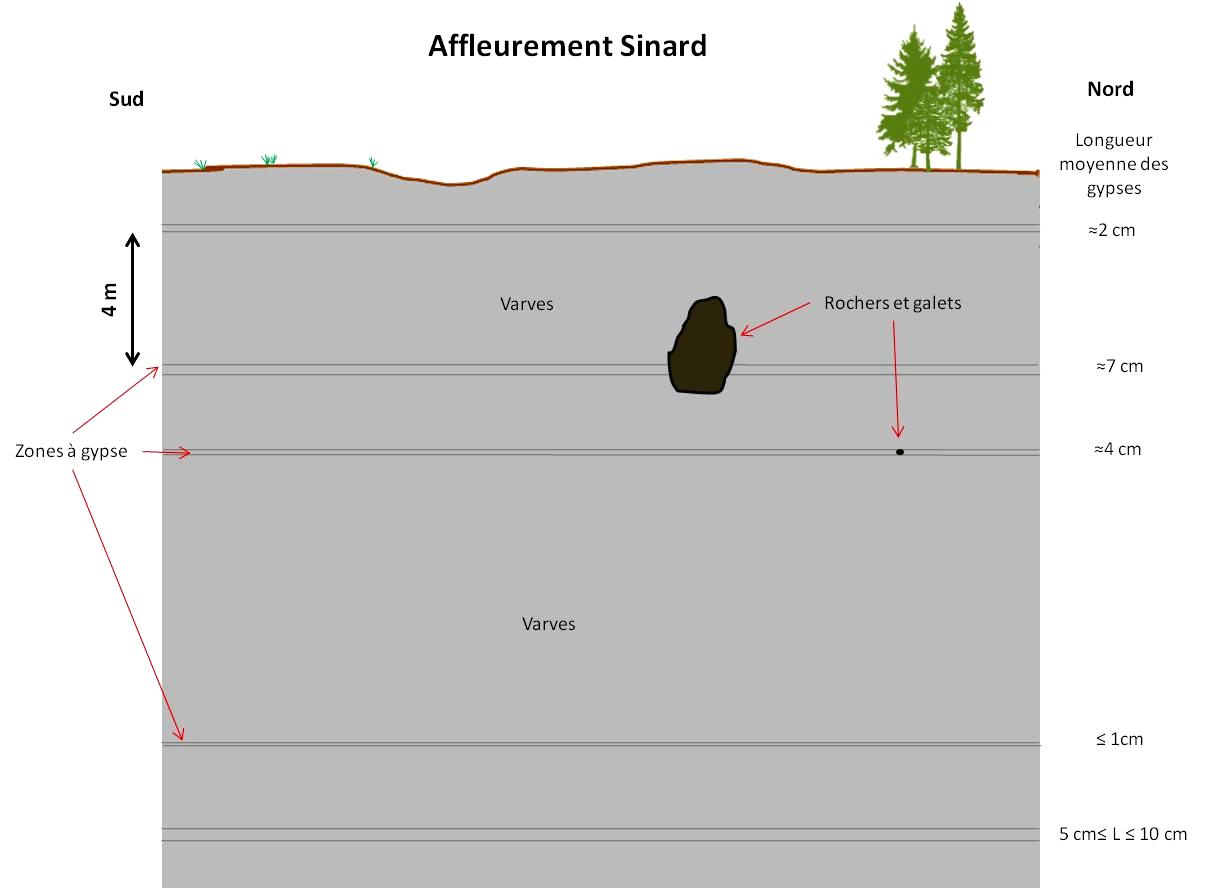
Dans les contreforts du Vercors, des argiles appelées varves contiennent de tels cristaux de gypse centimétriques (cf. Barailler 2025[4]). Ces cristaux de gypse sont présents au sein de certaines strates d'argile. Lorsqu'on observe la succession verticale de ces strates, on voit majoritairement des strates sans gypse visible, certaines avec des petits cristaux et plus rarement certaines avec de gros cristaux de plus de 20 cm (figure suivante). Quelques galets ou rochers peuvent également être présents, enchâssés dans ces argiles.
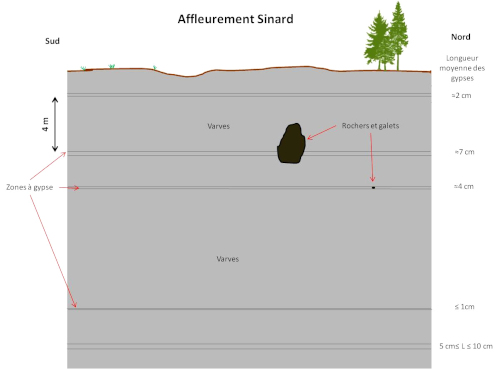
Source - © 2025 — Bernard Barailler
Des cristaux de gypse visible à l'œil nu sont présents dans certaines strates, les plus gros étant les plus rares.
Des couches de gypse sont connues pour se former par évaporation d'une saumure issue d'eau de mer ou de lac (évaporites). Le gypse remplit parfois des filons indiquant des circulations hydrothermales. Ici, le gypse n'est clairement ni évaporitique ni filonien. On peut donc se poser la question de l'origine et de la répartition de ces cristaux et chercher à comprendre comment ils se forment dans cet environnement. On se propose de comprendre pourquoi ils sont présents au sein de certaines strates sédimentaires et pas dans les autres, et pourquoi leur taille, homogène dans un niveau, est variable d'un niveau à un autre.
Par ailleurs, on observe des stries à l'intérieur de ces cristaux de gypse (figure ci-dessous) faisant penser à des stries de croissance. On s'intéressera alors au processus pouvant expliquer ces étapes de croissance[1]
.

Source - © 2025 — Bernard Barailler
Cristal de dimensions 5,5 × 2 × 1,5 cm. Les “stries” d'argiles marquant les étapes de croissance sont surlignées en tiretés rouges.
Répondre à toutes ces interrogations nécessite à la fois d'aborder quelques notions de cristallogenèse et aussi de remonter dans le temps à la période glaciaire du Würm (−45 000 à −20 000 ans).
Rappel sur le contexte du lac du Trièves, ses varves et ses gypses
Avant d'étudier la croissance des cristaux de gypse du Trièves, il faut bien rappeler le contexte géologique de leur matrice. En effet, ceci fixe les caractéristiques physico-chimiques qui vont présider à leur formation et à leur croissance : pression, température, pH, présence de matière organique ou de bactéries, présence d'autres ions, variations de concentration, etc.
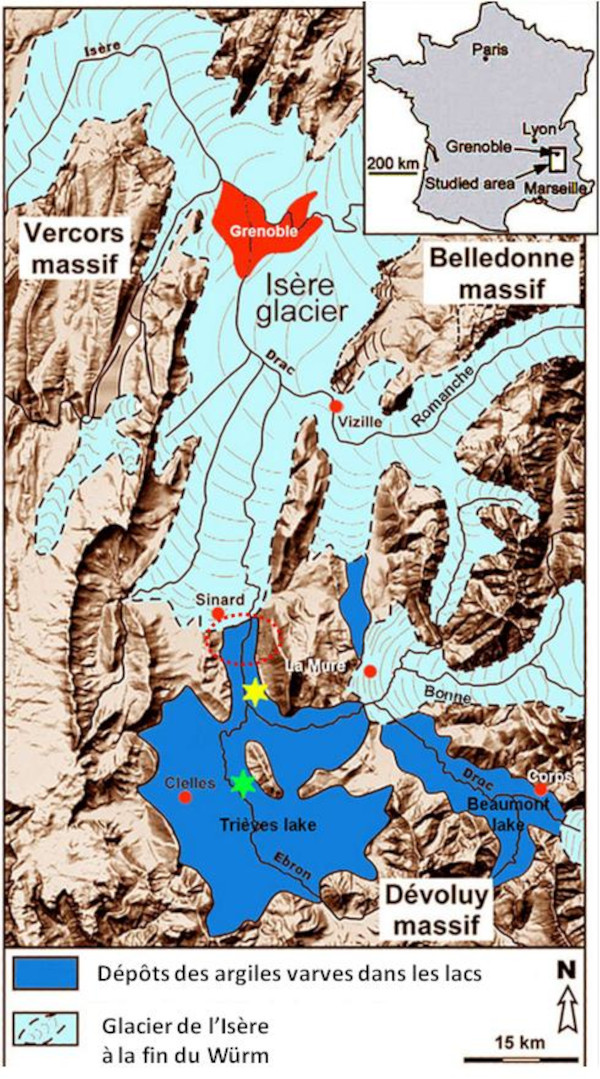
Les cristaux de gypse du Trièves sont enchâssés dans des argiles lacustres datant du Würm, l'une des périodes glaciaires pléistocènes des Alpes (cf. Thomas, 2021[30]). À cette époque, Sinard est au front Sud du glacier de l'Isère (Grenoble est sous les glaces) qui débouche sur le lac du Trièves (figure 4, Jongmans et al., 2009[14]). L'érosion des piémonts du Vercors et du Dévoluy apportent cycliquement des sédiments et notamment des argiles qui se déposent au fond du lac.
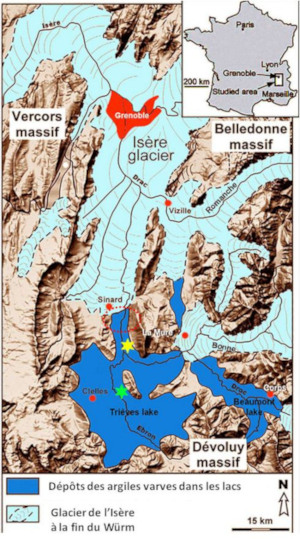
Source - © 2009 — D'après Jongmans et al. [ 14 ]
La zone de Sinard (cerclées de pointillés rouges) est située dans le lac du Trièves dans lequel arrivent plusieurs langues glaciaires, dont le glacier de l'Isère.
La formations et les caractéristiques physicochimiques des varves de Sinard ont été présentées dans un précédent article (Barailler, 2025[4]). Ces argiles varvées sont la matrice dans laquelle les gypses se sont développés. Le modèle classique de croissance cristalline est celui où les cristaux s'expriment dans des “vides” (“remplis” d'air ou d'eau). Ceci donne des filons ou bien des géodes. Ici, on est dans un modèle de croissance confinée (confined crystal growth) et particulièrement dans un environnement poreux, celui des argiles varvées.
L'observation de gypses (pluri)centimétriques, montre que les argiles qui sont au-dessus des cristaux ont subi une déformation plastique (figure 5). Les niveaux d'argiles sur lesquels sont “posés” les gypses sont moins déformés par la croissance des cristaux que les niveaux qui les recouvrent, certains, par endroits, étant même clairement “déchirés” par les gypses les plus gros (Barailler 2025[4]).

Source - © 2024 — Bernard Barailler
Les niveaux sur lesquels “reposent” les gypses sont moins déformés que les niveaux les recouvrant.
Sur cette image, les cristaux mesurent environ 1,5 cm de long. Les strates de varves qui recouvrent ces gypses à partir de leur moitié supérieure font 2 mm d'épaisseur en moyenne. On compte environ 8 à 10 strates déformées jusqu'à retrouver la planéité générale.
Comment se forme le gypse en général… et dans le Trièves
Dans le lac du Trièves, la croissance du gypse se réalise à partir d'ions en solution dans l'eau présente au sein des argiles varvées. Ce type de croissance “classique” à partir d'ions en solution a été étudiée dans l'article Comment se forment les cristaux ? Du bécher à la croissance hydrothermale (Barailler, 2024[3]).
Courbe de solubilité du gypse
La cristallisation du gypse correspond à la réaction suivante : 2 H2O + Ca2+ + SO42− ↔ CaSO4·2H2O. Les ions en solution se combinent pour former des cristaux de gypse.
À l'équilibre, on définit le produit de solubilité par Ks = a(Ca2+)eq × a(SO42−)eq × a²(H2O))eq, où “a” est l'activité chimique des différentes espèces chimiques et “eq” indique l'état d'équilibre (Barailler, 2024[3]). Pour une solution aqueuse, on peut écrire plus simplement KS = Ca2+eq × SO42−eq , où, par simplification d'écriture, Ca2+ représente l'activité en ions calcium, activité égale à sa concentration divisée par une concentration de référence (une activité chimique n'a pas d'unité), ce qui revient à prendre l'activité comme la “valeur” de la concentration (Dequincey, 2025[8]). Notons S, le produit S = Ca2+ × SO42− de la solution considérée.
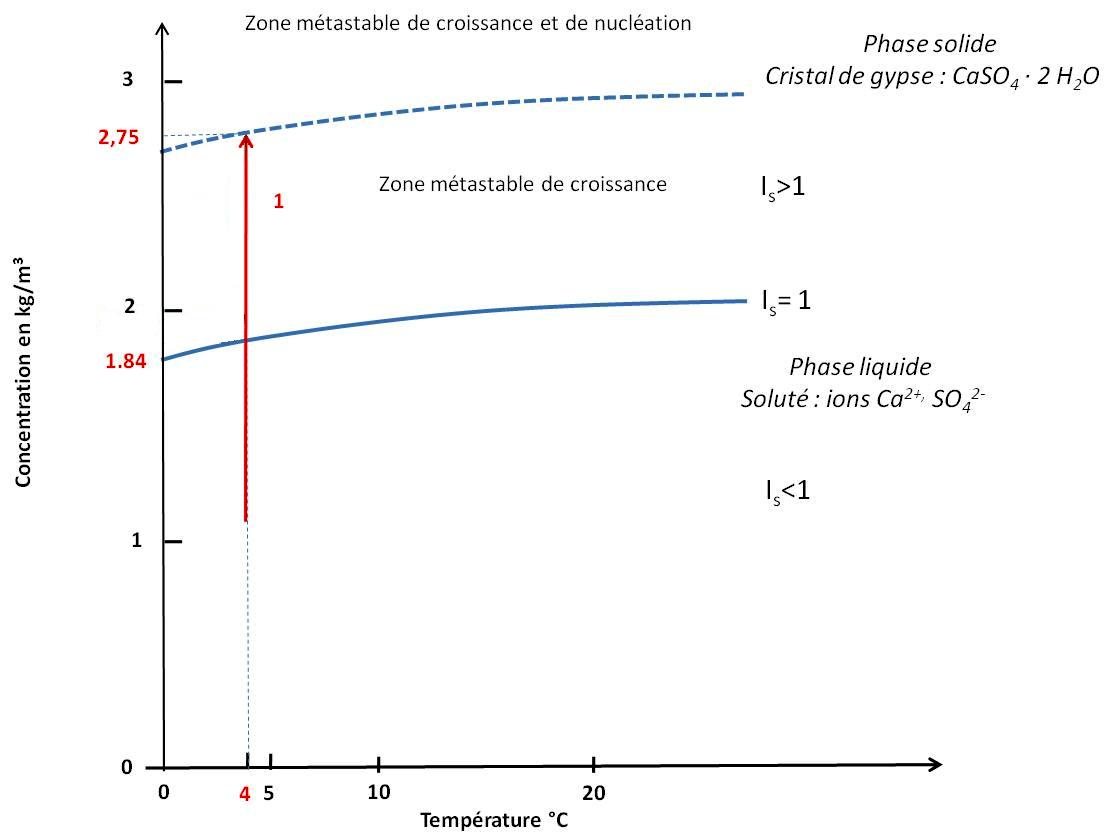
Plutôt que de donner la valeur de KS, on donne parfois un équivalent “quantité de minéral dissout” ou solubilité, qui correspond en fait à la quantité maximale de minéral qu'il est possible de mettre en solution dans de l'eau pure. Ainsi, pour le gypse, de masse molaire 172 g/L, un KS de 1,35.10−4 à 20°C et 1 atm correspond à une solubilité de l'ordre de 2 g/L (Dequincey, 2025[8]) (figure 6). L'index de saturation est défini par IS = S / KS (IS = 1 à l'équilibre) et permet d'exprimer aussi S en terme de “solubilité” (figure 6). La solubilité étant fonction de la pression et de la température, on peut représenter, par exemple, la courbe de solubilité du gypse en fonction de la température par rapport à laquelle on peut positionner la solution étudiée (figure 6).
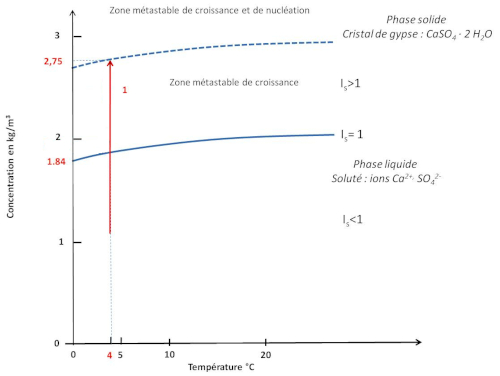
Source - © 2025 — Bernard Barailler d'après Pelser et al., 2003 [ 22 ]
Le trait rouge indique la position d'une solution aqueuse à 4°C.
Sur la figure ci-dessus, une solution à 4°C sous-saturée (IS < 1, S < KS) ne permet pas la cristallisation de gypse (par contre elle peut en dissoudre si du gypse est mis en contact). Lorsque IS = 1 (S = KS), on est à saturation (ici, à 1,84 g/L, soit 10,7.10−3 mol/L). À sursaturation, IS> 1 (S > KS), il peut y avoir formation de germes et croissance cristalline. La croissance cristalline consomme alors les ions en solution et le produit S diminue jusqu'à atteindre l'équilibre (S = Ks). On est dans la zone dite métastable de croissance. Un seuil de sursaturation maximal induit une nucléation spontanée au sein de la solution, et donc la croissance de cristaux de gypse jusqu'à revenir à saturation. Ce seuil de sursaturation permet de définir un produit de nucléation spontanée KN sur le modèle du produit de solubilité (ici, 2,75 g/L, soit 16,0.10−3 mol/L).
À partir d'une solution sous-saturée, il faut donc au moins atteindre la saturation, au plus le seuil de sursaturation, pour qu'une cristallisation et une croissance cristalline se développe.
Les conditions de formation des gypses du Trièves
Rappelons les conditions physico-chimiques associées à la formation de ces gypses, telles que décrites dans Barailler, 2025[4].
La présence de varves nécessite notamment une profondeur minimum de lac de 20 m. Dans le cas du lac du Trièves la profondeur va jusqu'à 200 m, et on peut estimer qu'au fond de ce lac, et dans la couche de dépôt sédimentaire, régnait une température de 4°C. Les variations de températures y sont minimes, elles peuvent contribuer à franchir la courbe de solubilité mais à priori de façon marginale (déplacement horizontal vers la gauche en cas de légère baisse de température par apport d'eaux de fonte au printemps ou en été).
On part d'une solution diluée, le lac alimenté par eaux de pluie, de ruissellement sur les versants des massif, de fonte de glacier… Cette solution contient à priori des ions calcium du fait que le glacier “circule” sur un substratum calcaire et que les massifs avoisinants sont aussi des massifs calcaires. Des sources ponctuelles de sulfures existent à proximité du lac (voir plus bas).
Les gypses se formant à l'intérieur des varves, leur “approvisionnement” en ions est directement lié au volume de solution accessible à un cristal en croissance. On a vu que, parallèlement aux strates de varves, la perméabilité horizontale est réduite : k//20°C = 10-8 m/s. Perpendiculairement aux strates, la perméabilité verticale l'est encore plus : kꓕ 20°C = 10-10 m/s. À 4°C, la perméabilité est encore plus faible kꓕ 4°C = 0.625 10-10m³/s (Barailler, 2025[4]). Toutefois des zones de drainage de fluide plus intense peuvent exister, à la faveur de galets qui déforment les strates ou bien de fissures générées par la pression de cristallisation des plus gros cristaux. Ceci permet d'alimenter encore un peu en ions la croissance du cristal à partir d'un volume plus important de solution “accessible”. Il semble tout de même y avoir une limite car on connait peu de cristaux dépassant les 20 cm de longueur dans le secteur de Sinard.
La question est donc de savoir par quel processus on passe d'une solution diluée contenant assurément des ions calcium et potentiellement des ions sulfate, à des conditions de cristallisation de gypse.
Les modèles “classiques” de cristallisation du gypse
Évaporation et évaporites. Le modèle le plus classique de genèse du gypse est celui des évaporites. L'eau s'évapore, la concentration en ions dans le fluide restant augmente jusqu'à franchir les courbes de saturation (cf. Barailler 2024[3], partie Les évaporites). Mais dans un lac glaciaire à 4°C, alimenté régulièrement en eaux pluviales et de fonte de glacier, ce modèle ne fonctionne pas.
Gel de saumure. Un autre modèle est basé sur la formation de glace (Geilfus et al., 2013[11]). Lors de la transition de phase liquide-solide (eau-glace) les ions restent essentiellement dans la fraction liquide, la glace étant appauvrie en solutés. Leur concentration augmente alors dans la solution résiduelle. Mais au niveau des sédiments à plus de 20 m de profondeur, l'eau ne gèle pas et reste à environ 4°C. Ce second modèle est donc lui aussi inopérant.
Altération et amas sulfurés.. Une troisième possibilité est la production de gypse à partir de l'altération d'amas sulfurés comme à Albas (Aude)(lien externe - nouvelle fenêtre). Cependant, dans les varves du Trièves, on ne trouve pas de traces de tels amas sulfurés. Les gypses sont répartis relativement uniformément sur les paléo-horizons. De plus, les sources de sulfure sont éloignées des varves (plus de 100 m de profondeur au minimum) (Matheson et Jones, 2015[20] ; Chen et al., 2018[????]).
Les modèles courants de formation de gypse ne pouvant apparemment pas s'appliquer à notre cas d'étude, un autre modèle basé sur une observation “industrielle”, est proposé.
Osmose inverse et concentration des solutions
Le gypse entartre les membranes de désalinisation
L'étude et la mise en œuvre de désalinisation de l'eau de mer montre que des dépôts de gypse ont lieu dans les membranes d'osmose inverse (Shaikh et al., 2015[29] ; Rahman, 2013[25] ; Abdel-Aal et al., 2015[1]). Cette formation de tartre sur les membranes d'osmose inverse (OI) est un problème majeur, car elle affecte les performances de la désalinisation. Le principal composé qui forme du tartre sur les membranes d'OI est le sulfate de calcium dihydraté (… le gypse !), suivi du carbonate de calcium. Dans le processus d'osmose inverse, il existe un flux de perméat (l'eau “désalinisée”) et de rejet. Dans le flux de rejet, les sels de l'eau d'alimentation sont concentrés par le processus. Si une sursaturation en sulfate de calcium dihydraté se produit et que la limite de solubilité (et/ou de sursaturation) est dépassée, alors le tartre se forme sur la membrane.
Les varves comme membranes filtrantes d'osmose inverse
Dans un fluide comme l'eau, la concentration des ions a tendance à s'homogénéiser par les phénomènes de diffusion, d'agitation thermique et de convection. Toutefois si on considère les varves comme des membranes semi-perméables, elles laisseraient passer les molécules d'eau et retiendraient les ions en solution dans leur porosité.
Les sédiments, lors de leur dépôt, contiennent beaucoup d'eau ainsi que des ions calcium et/ou sulfate. Au fur et à mesure que l'épaisseur des sédiments augmente, la pression devient plus forte. Il y a compaction, l'eau est chassée de la porosité des argiles considérées, ici, comme une membrane semi-perméable. Les ions ne peuvent alors pas la traverser et restent “piégés” dans la solution résiduelle de la couche compactée. Dans cette couche, la solution s'enrichit donc en ions (leur concentration augmente).
La différence de concentration des deux côtés de la “membrane” exerce une pression osmotique. À l'inverse, avec une pression sédimentaire ΔP qui dépasse la pression osmotique, on force l'eau à quitter le compartiment sous pression en dépit de l'augmentation de concentration en soluté qui s'y produit.
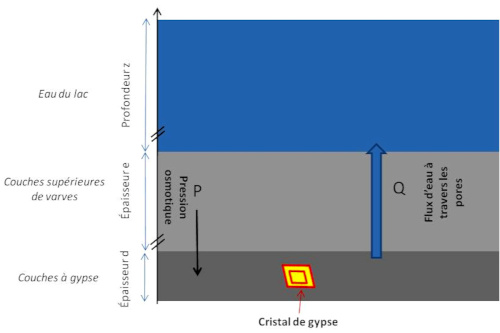
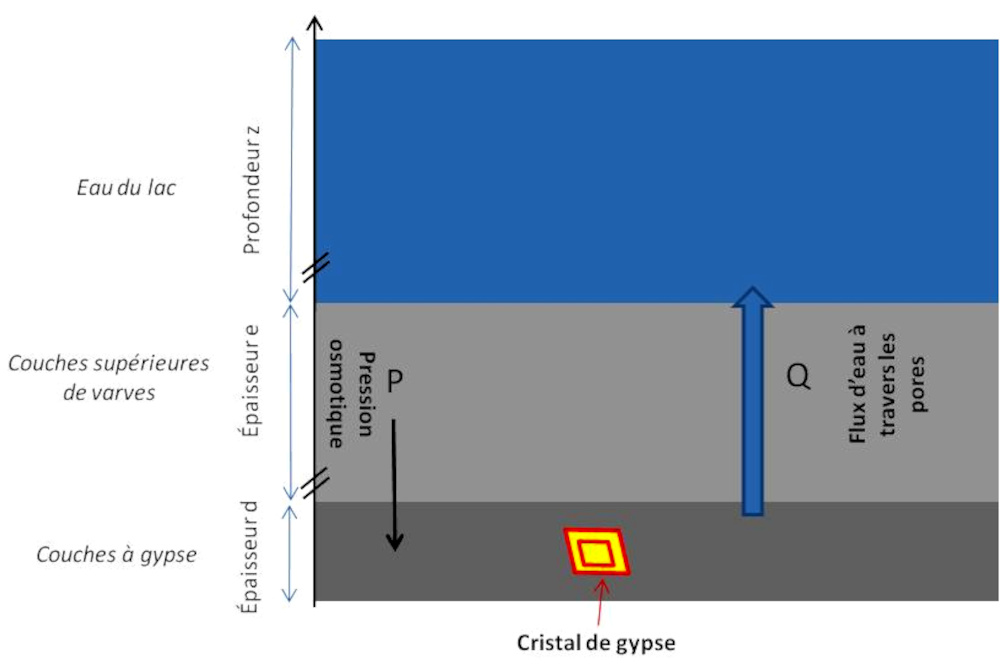
Source - © 2025 — Bernard Barailler
On se propose de modéliser la concentration de la solution présente dans la porosité des varves en considérant que les argiles se comportent comme une membrane semi-perméable entre la solution comprise dans une couche d’argile et l'eau du lac. La différence de concentration solution / lac (considéré comme un réservoir “constant”) crée une pression osmotique maintenue ou augmentée par la pression due au poids des sédiments sus-jacents.
Dans ce modèle nous prendrons la température de cristallisation à 4°C, celle supposée pour les conditions au fond du lac. La pression qui augmente la concentration en ions en chassant l'eau de la porosité permet de franchir la courbe de solubilité (figure 6). La cristallisation peut avoir lieu dès la saturation atteinte. Au maximum, la concentration atteint le seuil de nucléation spontanée auquel la nucléation s'opère et un cristal de gypse croît.
Afin de modéliser la nucléation nous prenons des hypothèses simplifiées.
Un germe à taille critique se développe au-dessus de la courbe de nucléation (figure 6). C'est une nucléation homogène spontanée.
L'action des autres ions est dans un premier temps considéré comme négligeable.
On est proche des conditions d'équilibre.
Application du modèle de membrane
Expression de la pression sédimentaire
Sous une épaisseur “e” de sédiments et une profondeur “z” du lac, la différence de pression entre la couche de varves considérée et le fond du lac est :
ΔP = (ρeau.g.z + ρargile.g.e) − ρeau.g.z = ρargile.g.e.
La mesure expérimentale de masse volumique pour une argile varvée de Sinard donne : ρargile ≈ 1733 kg/m3 (Barailler 2025[4]), une valeur estimée dans des sédiments lacustres équivalents à faible profondeur donne une valeur de l'ordre de 1170 kg/m³ (Loso, 2008[17]).
Expression de la pression osmotique
La loi de Van't Hoff exprime la pression osmotique pour les solutions réelles de la façon suivante :
Π.V = − R.T.ln(1-ϒ.x), où Π est la pression osmotique, V le volume molaire occupé par le solvant, R la constante des gaz parfaits (8,314 J.mol−1.K−1), T la température absolue en Kelvin, ϒ le coefficient d'activité du soluté et x la concentration du soluté.
Ici, nous prendrons un modèle de calcul simplifié de la loi de Van't Hoff(lien externe - nouvelle fenêtre) dont l'expression rappelle la loi des gaz parfaits :
Π = C.R.T d'où C = Π/ R.T, où C est la concentration du soluté en mol/m3.
Épaisseur de sédiments nécessaire pour compenser la pression osmotique d'une solution saturée ou sursaturée
Pour rappel, la réaction de cristallisation / dissolution est la suivante :
2 H2O + Ca2+ + SO42− ↔ CaSO4·2 H2O.
On pose X = [Ca2+] et Y = [SO42−]. Par définition, on atteint la saturation lorsque KS = X.Y, et la nucléation spontanée lorsque KN = X.Y (en considérant que les activités sont “égales” à la valeur des concentrations, voir plus haut).
En négligeant les ions autres que Ca2+ et SO42−, la concentration de la solution est alors C = X + Y et donc, à saturation C = X + KS / X (ou C = X + KN / X si on s'intéresse au seuil de nucléation spontanée).
En cas de flux d'eau possible, une concentration C n'est possible dans un niveau de varve que si la pression sédimentaire équilibre au moins la pression osmotique. À minima, on a donc ΔP = Π, soit ρargile.g.e = C.R.T, d'où e = (R.T/ ρargile.g).C, soit e = α.C, en posant α = (R.T/ ρargile.g).
On a alors la relation e = α.C = α.(X + K / X), avec X l'activité (“concentration”) de Ca2+ et K le produit de solubilité à saturation ou au seuil de nucléation spontanée, selon le cas envisagé.
On voit qu'épaisseur de sédiments et concentration de la solution sont proportionnelles. L'épaisseur minimale compensant la pression osmotique correspond donc à la concentration minimale en solution à saturation ou au seuil de solubilité. On peut rapidement montrer que la concentration totale de la solution est minimale lorsque X = Y = √KS (ou √KN, selon qu'on s'intéresse à la saturation ou à la nucléation spontanée), avec, de fait, C = 2.X (Dequincey, 2025[8]). À 4°C, les valeurs de solubilité de la figure 6 correspondent à des concentrations en solution CSat = 21,4.10−3 mol/L [KS = 1,15.10−4 et X = 1,07.10−2] ou CNuc =32,0.10−3 mol/L [KN = 2,56.10−4 et X = 1,6.10−2].
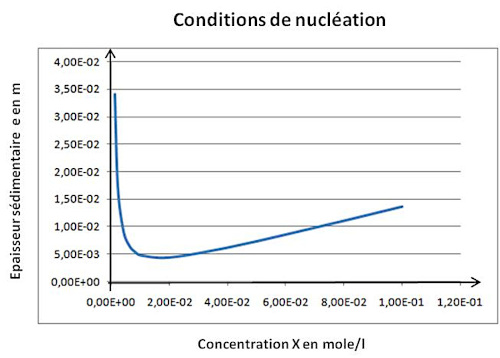
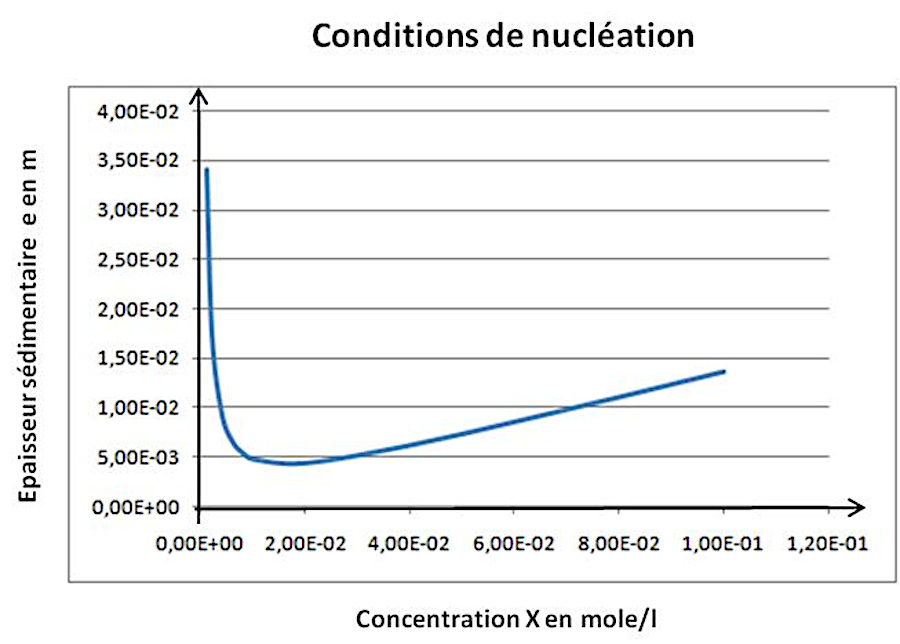
Source - © 2025 — Bernard Barailler
On a un minimum d'épaisseur de l'ordre de 5 mm dans les conditions “idéales” (X = Y, C minimale). Lorsqu'on s'éloigne de ces conditions, l'épaisseur de sédiments nécessaire pour compenser la pression osmotique augmente.
On a, en prenant g ≈ 10 m.s−2 (on cherche un ordre de grandeur) et avec T = 277 K, α = (8,134 × 277) / (1733 × 10) = 0,130. On a alors des épaisseurs de sédiments nécessaire de l'ordre de eSat = 21,4.10−3 × 0,130 = 2,8 mm et eNuc = 32,0.10−3 × 0,130 = 4,2 mm. On se rend compte que des épaisseurs très faibles sont suffisantes, épaisseurs pour lesquelles les argiles doivent être peu compactée. Ainsi, si l'on prend la valeur de masse volumique de 1170 kg/m³ mesurée par Loso (2008[17]) dans des varves actuelles à faible profondeur, on obtient α′ = 0,192 et des épaisseurs e′Sat = 4,1 mm et e′Nuc = 6,1 mm. On reste à des épaisseurs de l'ordre de 5 mm pour une pression sédimentaire compensant une solution de concentration minimale à saturation, sursaturée, voire au seuil de nucléation spontanée.
Estimation du temps nécessaire pour atteindre les conditions de nucléation spontanée
L'épaisseur des couches annuelles est variable en fonction des apports sédimentaires. Dans la zone du Trièves, elle est comprise entre 1 mm et 10 cm (Barailler, 2025[4]). À Sinard, l'épaisseur annuelle mesurée est d'environ 2 mm. Il suffirait donc de seulement 2 ou 3 ans pour faire un premier germe de gypse (… dans les conditions stœchiométriques “idéales”).
Remarques, limites et questions liées au modèle
Cette modélisation est très simplifiée, elle permet de montrer que la formation de gypse à partir d'une solution concentrée par osmose inverse est à priori envisageable. Elle donne également un ordre de grandeur de l'épaisseur de sédiments nécessaire compatible avec l'analyse de l'affleurement de Sinard.
Une faible influence de la pression sur la solubilité
On n'a pas pris en compte la variation de solubilité liée à la pression car on reste à basse température et sous de faibles pressions (même dans des sédiments au fond d'un lac de 200 m), et même si des mesures et des modèles de solubilité du gypse montrent que vers 4°C la solubilité du gypse augmente de l'ordre de 50-60 % lorsqu'on passe de 1 bar à 500 bar (Yuan et al., 2025[32]), puisque, même en se plaçant à 20-22 bar, les calculs donneraient les mêmes ordres de grandeur (épaisseur sédimentaire minimale infracentimétrique).
Porosité résiduelle et facteur de concentration par osmose inverse
Les varves de Sinard ayant une porosité de l'ordre de 40 % (Giraud et al., 1991[12]), on en déduit un facteur de concentration maximal d'un soluté par “expulsion” du solvant (l'eau). En effet, en considérant, à l'extrême, les dépôts initiaux de varves comme une solution d'argiles en suspension dans de l'eau avec une “porosité” de quasi 100 %, la porosité résiduelle de 40 % indique que le volume d'eau a été divisé par 2,5 et donc, par conséquent, que la concentration de chaque soluté a pu être multipliée par 2,5. Le produit final X.Y peut donc aboutir à une valeur de saturation ou de nucléation spontanée après concentration avec des valeurs initiales chacune 2,5 fois plus faibles et donc un produit 2,5 × 2,5 = 6,25 fois plus faible, soit, par exemple un produit X.Y initial égal à 1,8.10−5 pour aboutir au final à la (sur)saturation à 4°C (KS = 1,15.10−4).
Des concentrations initiales réelles différentes des conditions “idéales”
Les calculs pour la solution présente au sein des varves ont été menés avec la concentration minimale Cmini = 2√K. Il est plus que probable que les concentrations initiales en calcium et sulfate n'étaient pas égales. Prenons une teneur en calcium supérieur à celle en sulfate. À saturation / nucléation, si X = μ.√K alors il “suffit” que Y = 1/μ.√K, et la concentration C devient alors égale à (μ + 1/μ).√K soit C = 1/2(μ + 1/μ).Cmini. Très rapidement, dès que μ dépasse 10, C ≈ μ/2.Cmini. Dans ce cas, il faudra une pression sédimentaire aussi μ/2 fois plus élevée pour compenser la pression osmotique. En prenant aussi en compte le fait que la solution initiale comprend d'autres solutés (Mg2+, Na+, Cl−…), pour une solution à saturation 200 fois plus concentrée que la solution “idéale” du modèle, l'épaisseur de sédiments à envisager est 100 fois plus importante que l'épaisseur minimale estimée… soit de l'ordre de 50 cm. On reste dans des conditions très proches de la surface.
Une compaction maximale rapidement atteinte
Si on reste sur un modèle de concentration par osmose inverse, cette concentration ne peut se développer que tant que les varves se compactent. Une étude dans des varves d'un lac suédois à dépôts annuels de l'ordre de quelques millimètres à un centimètre (Maier et al., 2013[18]) montre que la porosité décroit rapidement avec la profondeur. La porosité minimale est presque atteinte au bout d'une trentaine d'années, même si une légère compaction est encore possible. On est donc sur une épaisseur maximale de compaction par baisse de porosité de l'ordre de 50 cm environ. Ceci montre encore que la concentration envisagée n'est effective que dans le premier mètre, et même moins, ce qui renforce l'idée de cristallisation proche de la surface.
Une croissance vers le haut plus importante que vers le bas
Un autre argument en faveur de la cristallisation à faible profondeur est l'observation de la croissance différentielle des cristaux vers le haut et vers le bas (figure 5). Statistiquement, en mesurant la croissance d'une série de cristaux de gypse pluricentimétriques, il apparait que le développement vertical vers le haut, par rapport à la couche de référence, est environ deux fois plus important que le développement vers le bas. Si la force de cristallisation (cf. Barailler, 2025[4]) explique sans problème la capacité des cristaux à croitre en déformant les varves, une différence de “résistance” à cette croissance vers le haut et vers le bas doit expliquer les observations. À faible profondeur, on peut envisager une différence de plasticité générale vers le haut et vers le bas du fait de la moindre compaction des couches au-dessus du niveau de gypses par rapport à celle des couches en dessous. On a vu (Maier et al., 2013[18]) qu'au-delà d'un mètre de profondeur, voire moins, la porosité est “fixe”. À plus forte profondeur, le poids des sédiments augmente progressivement et limite une éventuelle “résistance” différentielle verticale.
Quelques questions restantes
Envisageons maintenant l'environnement du lac de Trièves et étudions les disponibilités en calcium et sulfate pour expliquer la possibilité de formation de gypse mais aussi pour tenter de comprendre la restriction de la présence de gypse à certains niveaux seulement.
Nous nous intéresserons ensuite à la croissance par étapes des gypses, qui semblent marquer des arrêts et des reprises de cristallisation, pour voir comment intégrer cette observation dans le modèle.
Enfin, nous passerons en revue l'effet des autres solutés généralement présents dans les eaux de lac sur la cristallisation du gypse, en considérant tout particulièrement la teneur en sel NaCl.
Provenance des ions calcium et sulfate dans le lac du Trièves
Des précipitations atmosphériques aux océans, les ions se concentrent. Le tableau suivant compile les moyennes mondiales des concentrations des eaux de pluies, de rivières et marines, et les concentrations mesurées dans les grands lacs d'Amérique du Nord.
Tableau 1. De l'eau de pluie à l'océan, teneurs en ions calcium et sulfate
| Milieu | [Ca2+] mol/L | [SO42−] mol/L | Produit des activités calcium×sulfate |
|---|---|---|---|
| Précipitations atmosphériques | 7,8.10−6 | 1,4.10−5 | 1,1.10−10 |
| Fleuves et rivières | 3,4.10−4 | 9,1.10−5 | 3,1.10−9 |
| Lacs d'Amérique du Nord | 7,7.10−4 | 5,2.10−4 | 4,0.10−7 |
| Océans | 1,0.10−2 | 2,9.10−2 | 2,9.10−4 |
En plus des concentrations moyennes en Ca2+ et SO42−, le produit des “activités” est donné pour comparaison au produit de solubilité du gypse (KS = 1,35.10−4 à 20°C ou 1,15.10−4 à 4°C) et au produit d'activité minimum dans la solution initiale pour atteindre la saturation à 4°C après compaction des varves dans le modèle proposé, X.Y = 1,8.10−5.
D'après Probst et Probst (2015[24]), Chapra et al. (2012[5]), Waker et Robarts (1995[31]).
On voit que malgré la concentration progressive des ions de la pluie aux lacs, les teneurs sont insuffisantes pour atteindre le seuil de saturation dans les eaux continentales ou même seulement le seuil de “pré-concentration” estimé plus haut. Même en prenant des données sur différents environnements lacustres (Larsen et al., 1997[16]), on n'arrive pas au seuil désiré.
On notera ici la sursaturation des océans, dans lesquels le gypse ne précipite pas spontanément, ce qui montre bien qu'il ne suffit pas de s'intéresser aux seules teneurs en ions calcium et sulfates mais que la composition globale a une influence certaine sur la précipitation effective des cristaux.
Les bassins versants du paléo-lac du Trièves sont largement calcaires et les varves contiennent entre 15 et 50 % de carbonate de calcium, la teneur en ions Ca2+ devait donc être “importante” mais aussi, et surtout, relativement stable dans cet environnement carbonaté. Si le gypse n'est pas trouvé dans tous les niveaux de varves, c'est donc certainement dû à la variation de la teneur en sulfate et/ou à des facteurs inhibiteurs de nucléation/précipitation. La teneur en ions sulfate des eaux continentales est très variable, elle dépend essentiellement de la géologie des bassins versants.
Les sulfures dans les bassins versants du lac du Trièves
Rappelons que contrairement à d'autres gisements comme Albas dans l'Aude, on ne trouve pas de trace de nodules pyriteux dans les varves de Sinard. Les ions sulfate ne semblent donc pas venir d'une décomposition in situ qui pourrait alimenter progressivement la concentration des ions jusqu'à saturation.
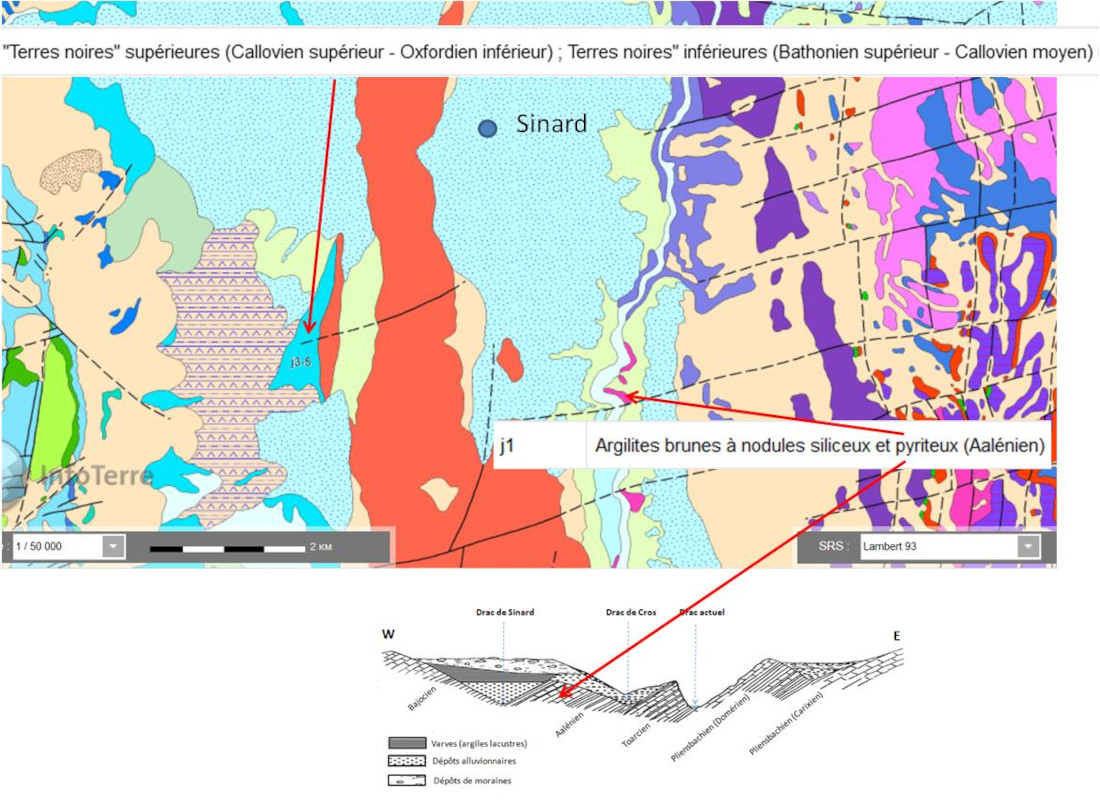
À l'époque du Würm, les ions sulfate du lac du Trièves proviennent probablement du lessivage et de l'oxydation des sulfures des Terres noires sous-jacentes (figure 09, j3-5 Bathonien à Oxfordien) (Thomas, 2021[30]). Mais le paléo-lac est situé sur des terrains aaléniens présentant des nodules pyriteux, terrains formant aussi certains abords du lac (figure 9, j1) sur lesquels “passent” des eaux d'alimentation.
Enfin sur le bassin du versant Est, le Dôme de Mure est un bassin houiller riche en sulfures.
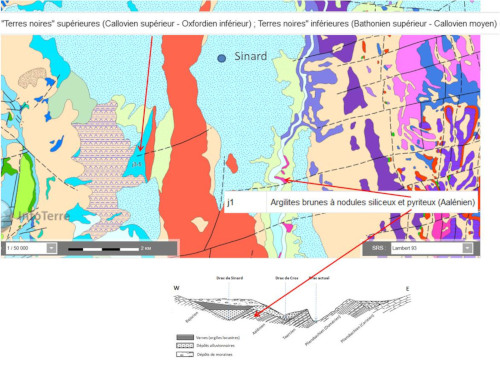
Source - © 2025 — D'après BRGM / InfoTerre
D'autre part, le Trias sous-jacent, évaporitique dans la région, parait peu affleurant à l'époque. L'unité géologique la plus ancienne mentionnée dans les coupes de la zone est le Pliensbachien. Deux séries géologiques la séparent du Trias. Toutefois quelques failles traversent la zone. Des remontées d'eau en provenance des couches sous-jacentes du Trias ne sont pas à exclure. En termes de proximité, leurs apports semblent probablement plus faibles. Une analyse isotopique de ces gypses pourrait permettre de trancher entre une origine “triasique profonde” et une origine “oxydation de surface” des sulfures. La présence des évaporites du Trias peut cependant jouer un rôle dans l'apport d'ions Na+ qui participent notablement à la nucléation du gypse.
Les bactéries et l'oxydation de la pyrite
L'oxydation de la pyrite en milieu acide est donnée par les équations suivantes.
(i) 2 FeS2 + 7 O2 + 2 H2O → 2 Fe2+ +4 SO4 2− + 4 H+,
(ii) 4 Fe2+ + O2 + 4 H+ → 4 Fe3+ + 2 H2O,
(iii) FeS2 + 14 Fe3+ + 8 H2O → 15 Fe2+ + 2SO42− + 16H+.
Ces processus chimiques sont considérés comme lents. Ils peuvent être accélérés par des bactéries comme Acidithiobacillus ferrooxidans. La bio-oxydation résultante multiplie par 250 la formation des ions ferriques Fe3+ (équation (ii) ci-dessus) (Pisapia, 2006[23]).
D'autres bactéries dites sulfo-oxydantes peuvent produire des sulfates selon les réactions suivantes.
(iv) n CO2 + 2n H2S + hν → CnH2nOn + n H2O + 2n S,
(v) S2− + 2 O2 → SO42−.
La réaction (iv) est réalisée en présence de lumière (hν) par le groupe des bactéries pourpres sulfureuses ou bactéries phototrophes sulfo-oxydantes. La réaction (v) est conditionnée à la présence d'oxygène (cf., par exemple, Selosse et al., 2008[28]). On peut envisager cette production de sulfates dans des zones lacustres peu profondes et riches en sulfures. Par contre, dans le fond du paléo-lac du Trièves, par 200 m de fond, il n'y a ni lumière ni oxygène, pas plus qu'au sein des varves par la suite. Le rôle catalyseur de ces bactéries peut donc être envisagé seulement le long des berges du lac où la profondeur est faible. Au fond du lac, si des sulfures étaient présents, leur décomposition est principalement d'origine chimique.
Rythmicité et durée de croissance des gypses de Sinard
Une croissance par étapes
À Sinard, on observe une grande variabilité de l'épaisseur des varves (Barailler, 2025[4]). Les couches de varves associées à la croissance des gypses sont celles qui enserrent le cristal (figure 10). Leur épaisseur est de l'ordre de 2 mm pour la partie claire printemps-été et infra-millimétrique pour la couche sombre automne-hiver. Le nombre de couches annuelles supérieures qui sont déformées jusqu'à l'englobement final des cristaux est de l'ordre de 8 à 10 (figure 10). On retrouve un nombre relativement analogue de fantômes de croissance dans les monocristaux (figure 3).

Source - © 2024 — Bernard Barailler
Ce cristal mesure environ 5,5 × 2 × 1,5 cm.
Les fantômes de croissance sont des dépôts d'argiles. Des expériences menés par Cody et Cody (1988[7]) avec de la bentonite montrent des résultats analogues : « Des coupes minces de cristaux de gypse cultivés expérimentalement […] montrent des fractures avec de fines inclusions d'argile le long de plans perpendiculaires aux axes c du gypse. » Ces fantômes d'argile pourraient marquer des arrêts de la croissance cristalline pendant lesquels de fines argiles peuvent se déposer sur et s'insinuer dans les anfractuosités du cristal. Mais la cristallisation reprend et s'arrête de nouveau plusieurs fois. Voyons quels sont les processus et les durées envisageables pour cette croissance par étapes.
Intégration du processus par étapes dans le modèle proposé
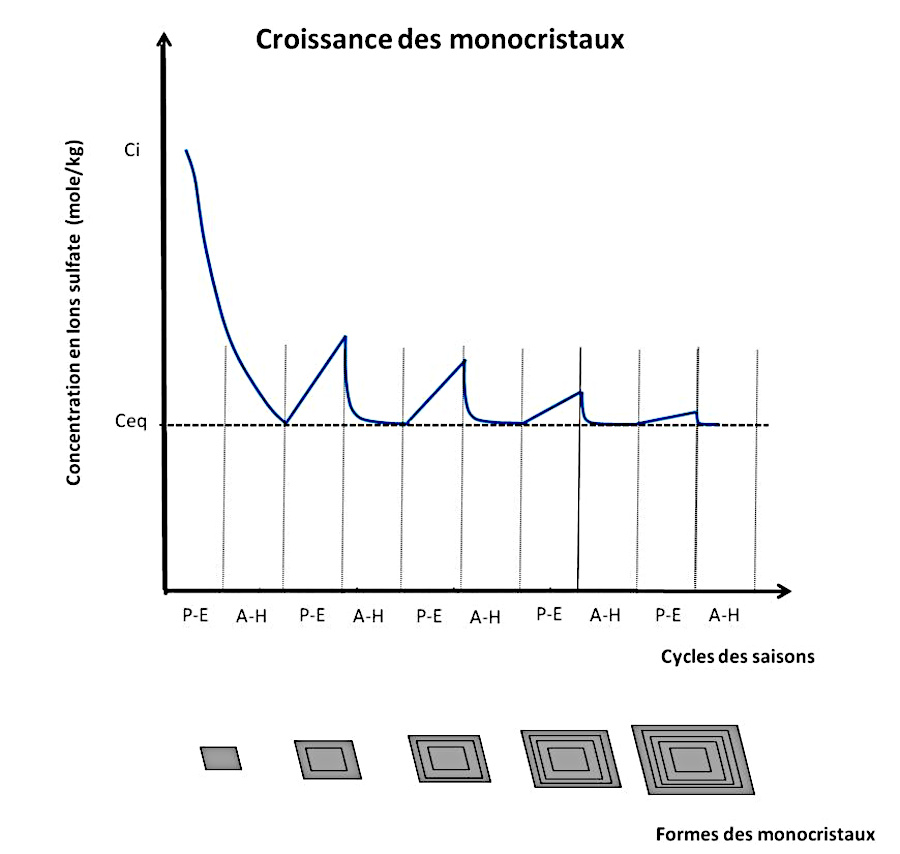
La figure 11 illustre l'enchainement de processus proposé pour expliquer non seulement la formation de gypse mais aussi, ensuite, sa croissance par étapes.
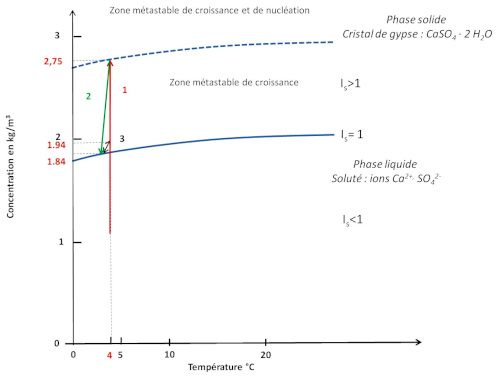
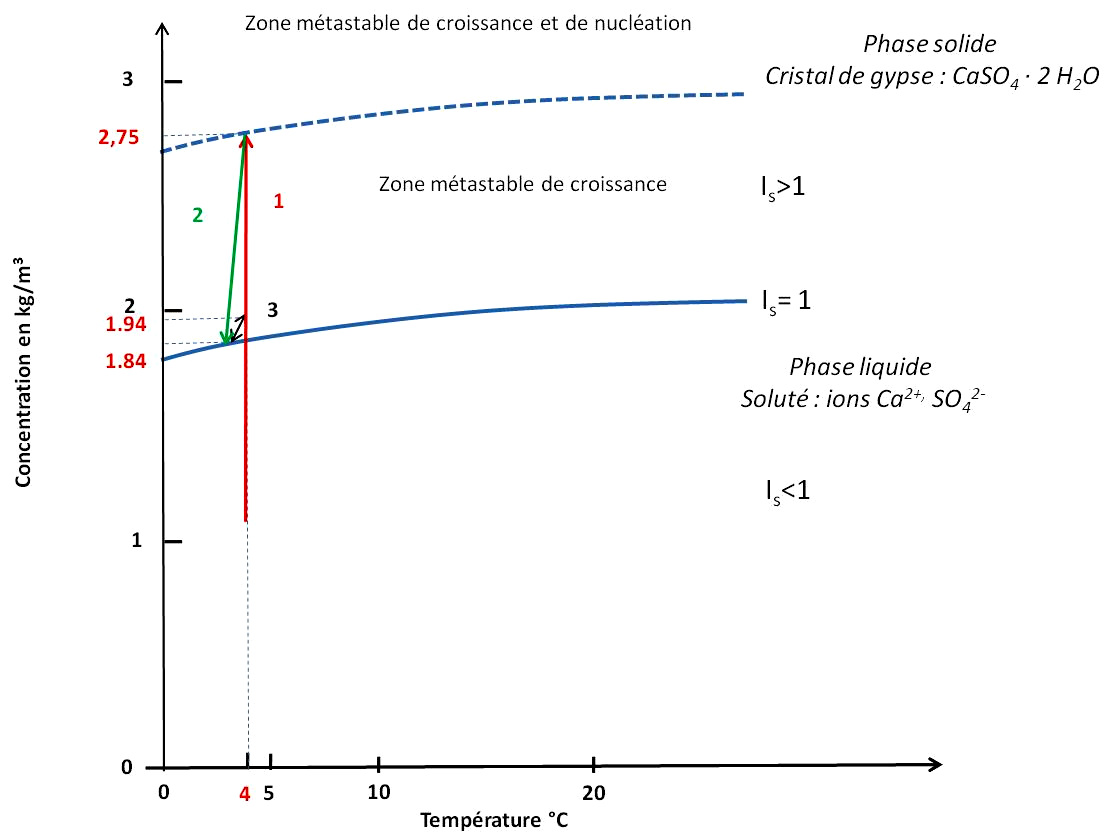
Source - © 2025 — Bernard Barailler
Pour des raisons de lisibilité, une légère baisse de température (2) et une sursaturation (3) de 0,1 g/L sont considérées. Le produit d'activité est représenté en équivalent solubilité du gypse en g/L.
Après la phase de nucléation (chemin 1, en rouge), la première croissance du cristal se produit jusqu'à la redescente à saturation, à 1,84 g/L (chemin 2, en vert). En effet le premier germe va pouvoir se développer dans la zone métastable de croissance en consommant les unités de croissance (ions et molécules d'eau) proches de lui. La consommation des unités de croissance fait baisser les concentrations jusqu'à revenir à la saturation “simple”. On peut également imaginer une légère baisse de température associée à la période printemps-été. En effet, durant l'hiver le lac est très certainement gelé et la glace constitue alors une barrière “opaque” au rayonnement du corps noir et contribue à garder la chaleur. À contrario, durant le printemps et l'été, un apport d'eau de fonte froide arrive, et descend vers le fond du lac.
Arrivé à l'équilibre, la croissance est interrompue. Au niveau microscopique, des dépôts de particules fines peuvent se produire sur les faces et les anfractuosités du cristal, formant des ombres de croissance. Pour que la croissance reprenne, une sursaturation est nécessaire. Comme le dépôt d'argiles est très faible en hiver, aucune évolution notable des concentrations n'est à envisager au sein des sédiments. Par contre, les dépôts sont plus importants pendant la période printemps-été. Chaque dépôt annuel, principalement en printemps-été, induit une augmentation de pression sédimentaire et donc une légère compaction, d'où une augmentation de concentration de la solution dans les varves affectées par osmose inverse (chemin 3, en noir, vers le haut). La croissance peut alors reprendre jusqu'à retour à l'équilibre (chemin 3, en noir, vers le bas). Ceci amène à une croissance “cyclique” liée aux dépôts annuels de varves. La concentration oscille alors entre la valeur d'équilibre/saturation et une sursaturation liée aux dépôts de l'année (chemin 3, en noir). La croissance cristalline s'arrête définitivement dès que la sursaturation n'est plus possible par arrêt de la compaction. On ne peut pas parler de possible épuisement de l'un des réactifs. En effet, la “consommation” de calcium peut être compensée par la dissolution de calcaire et la présence d'ions sulfate résiduels est certaine puisque sinon cela entrainerait la dissolution de gypse jusqu'à retour à l'équilibre (et donc à la présence de calcium et de sulfates dans la solution). Cependant, dans le contexte géologique du Trièves, on peut parler des ions sulfate comme facteur “limitant” (par rapport au calcium “abondant”).
Durée et atténuation de la croissance cristalline
Le mécanisme proposé implique à priori que les strates de croissance des gypses sont des strates plus ou moins annuelles. Ainsi, comme les gypses d'environ 1,5 cm d'épaisseur montrent moins d'une dizaine de strates, cela indiquerait une croissance en moins de 10 ans. De plus, comme la compaction des niveaux de varves décroit très rapidement avec la profondeur et donc avec le temps (Maier, 2013[18]), on peut penser que les sursaturations successives sont de moins en moins importantes (à surcharge sédimentaire équivalente).
La figure suivante (figure 12) propose un modèle simple des “cycles” de croissance des gypses de Sinard. Le schéma illustre l'évolution de la saturation en gypse en fonction du temps à partir de la formation d'un premier germe. En dessous, les dessins schématisent les stades de croissance des monocristaux avec leurs ombres de croissance.
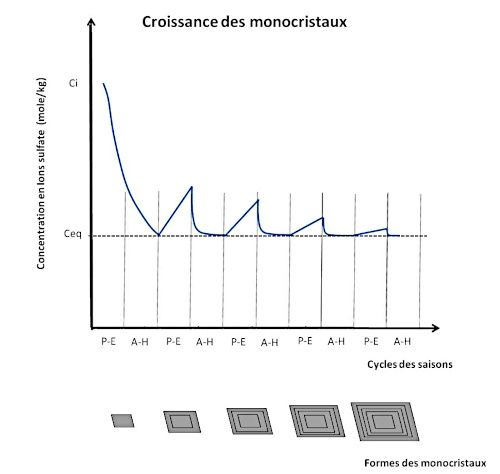
Source - © 2025 — Bernard Barailler
La taille du cristal formé à la première étape dépend, entre autres, du seuil de sursaturation ayant entrainé la nucléation. Les sursaturations ultérieures représentées sont des sursaturations certainement virtuelles qui indiquent en réalité la sursaturation maximale équivalente si la cristallisation avait lieu uniquement en fin d'épisode de compaction. À priori, la croissance a lieu dès que la saturation est à peine dépassée, sauf à estimer que les ombres de croissance induisent un léger palier énergétique nécessitant un certaine sursaturation pour que la cristallisation reprenne.
Durant la période printemps-été (PE), un apport de sédiments grossiers est fourni. La pression sédimentaire augmente alors. La température peut aussi légèrement baisser par un apport d'eau plus froide. Lorsque la pression sédimentaire augmente, l'eau de la solution mère résiduelle emprisonnée dans la couche où baignent les cristaux sort par les pores, ce qui entraine une augmentation des concentrations en ions sulfate et calcium. La croissance cristalline peut reprendre dans la zone métastable jusqu'à retour à l'équilibre cristal-solution. Durant la période automne-hiver (AH) le lac est gelé, l'apport sédimentaire est limité au fond du lac, la croissance s'arrête et des particules fines se déposent, formant les fantômes de croissance.
Nombre et taille des cristaux : la bataille pour les ressources
La nucléation en milieu confiné
Il est à noter que la réaction de nucléation du gypse pourrait démarrer rapidement de façon hétérogène à la surface de nano- ou microparticules (100 nm) de poussières. (Oshchepkov et al., 2020[21]). Un exemple de la taille moyenne des grains d'une varve est de l'ordre de 6 à 7 μm (Lapointe, 2012[15]).
Il peut paraitre difficile qu'un cristal se développe dans un univers confiné comme une argile. Cependant, quand on analyse les tailles des différents objets (tableau 2), on s'aperçoit que les ions solvatés sont plus de 2 000 fois plus petits que la taille des pores présents dans les argiles dans lesquels ils sont contenus. La valeur de la taille des pores est très variable, ici une valeur moyenne faible est donnée à titre d'exemple.
Tableau 2. Diamètres comparés de la molécule d'eau, de solutés, de pores et de germes cristallins
| Molécule, ion ou structure | Diamètre (nm) |
|---|---|
| Molécule d'eau | 0,343 |
| Ion calcium | 0,200 |
| Ion calcium solvaté | 0,824 |
| Ion sodium solvaté | 0,716 |
| Ion sulfate | 0,580 |
| Ion sulfate solvaté | 0,758 |
| Pore dans l'argile | 2000 |
| Germe cristallin (200 mailles élém.) | 6 |
Remarquons que la taille des molécules d'eau est plus faible que celle des autres solutés solvatés mais que la taille des petits pores envisagés reste bien supérieure. Toutefois, cela n'empêche pas d'envisager un processus de type osmose inverse car ce n'est pas la taille des pores qui importe et fixe la perméabilité, mais la taille des “connexions” entre les pores. De plus, le caractère “chargé” des ions peut retarder, limiter, voire empêcher leur migration du fait d'interactions avec les argiles.
D'après Marcus, 1998[24] ; Gaidis, 2020[9] ; Guillot et al., 2002[13].
Pour fixer les idées, un germe cristallin composé d'environ 200 mailles élémentaires aurait un diamètre de l'ordre de 6 nm (paramètres de la maille conventionnelle du gypse – mindat.org(lien externe - nouvelle fenêtre) – a = 5,68 Å, b = 15,20 Å, c = 6,50 Å). Imaginez une maison de 6 m de haut dans espace de 2 km de diamètre !
La nucléation est encore mal comprise dans ces détails. Toutefois les ordres de grandeur évoqués montrent qu'un germe pourrait se développer de façon hétérogène sur une nanoparticule au sein d'un pore d'une argile. Une fois rempli l'espace du pore, la force de cristallisation permet de “jouer des coudes” et de développer le cristal progressivement au sein des strates d'argiles (Barailler 2025[4]).
Une nucléation hétérogène à partir des nanoparticules d'argile est probable. La nucléation hétérogène est plus rapide que la nucléation homogène car pour le même nombre d'entités moléculaires, l'énergie de surface est plus faible. (Rong, 2018[27]). La nucléation secondaire, c'est-à-dire ici la formation d'un nouveau germe sur un cristal de gypse déjà formé, se produit généralement à une sursaturation beaucoup plus faible (Garside et al., 2003[10]).
L'influence du sel NaCl et du magnésium Mg2+
Afin de simuler la croissance du gypse dans les argiles, des expériences ont été menées sur des gels de bentonite (mélange argiles et eau) (Cody et Cody, 1988[7]). Les auteurs ont modifiés les paramètres de salinité, d'acidité et d'apport de matière organique afin de reproduire les conditions terrestres.
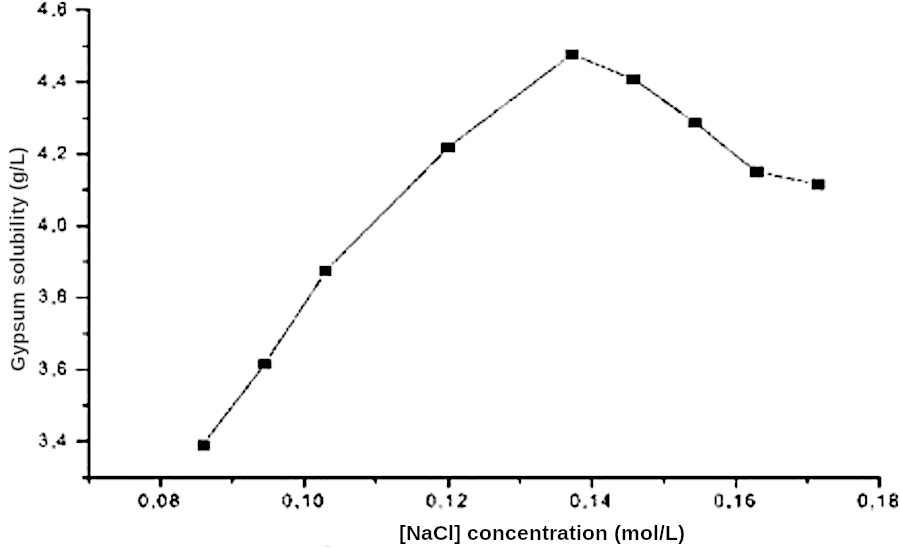
Un apport en sel augmente la solubilité du gypse et donc retarde d'autant sa cristallisation (figure 13). De plus, la cristallisation du gypse dans des solutions de chlorure de sodium a été étudiée en présence d'ions magnésium. Les ions Mg2+ ont un effet inhibiteur sur les deux étapes de précipitation : la nucléation et la croissance du gypse.
Les ions magnésium peuvent être inclus dans les sites octaédriques de l'illite qui est fortement présente dans les varves de Sinard.
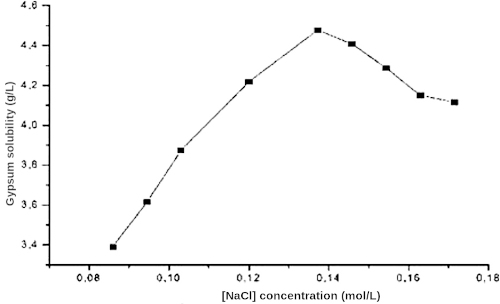
Source - © 2014 — Ben Ahmed et al. [ 2 ]
Ceci est cohérent avec les dépôts des évaporites car la dolomite [CaMg(CO3)2] précipite avant le gypse. Les ions sont en compétition pour capter les molécules d'eau nécessaires à leur solvatation et ensuite les relâcher lors de leur incorporation dans le cristal (Barailler 2024[3]).
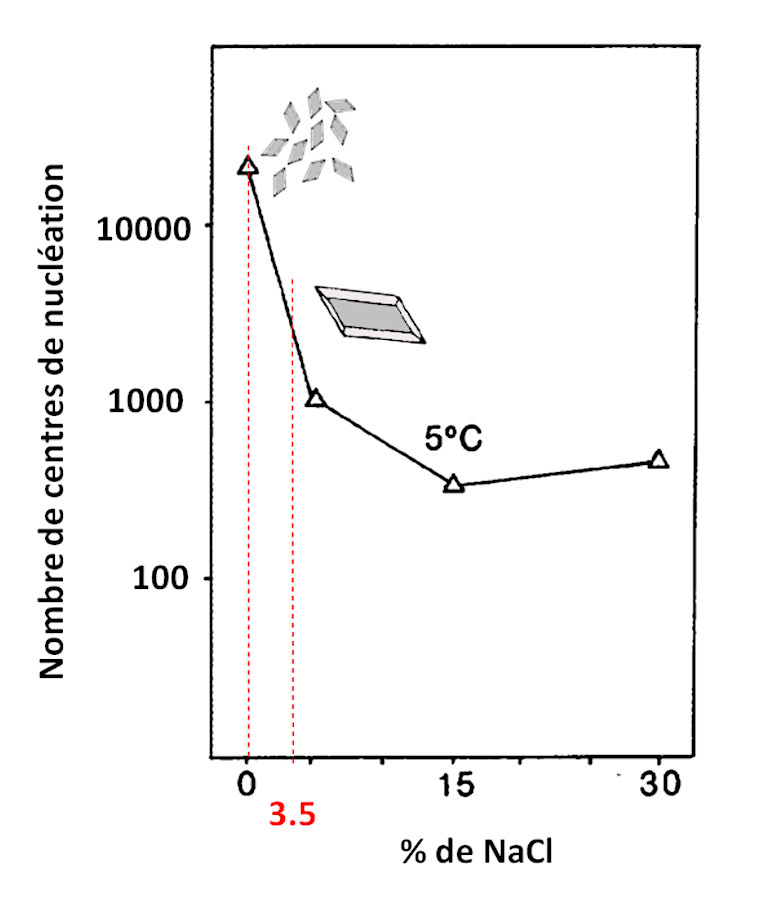
La figure suivante (figure 14) montre l'influence du sel (NaCl) sur la nucléation des cristaux de gypse. Il est à noter que lors de ces expériences 0,1 % d'acide tannique a été ajouté afin de mieux simuler l'apport de matières humiques terrestres dans un lac. Le pH a été maintenu vers 7,5.
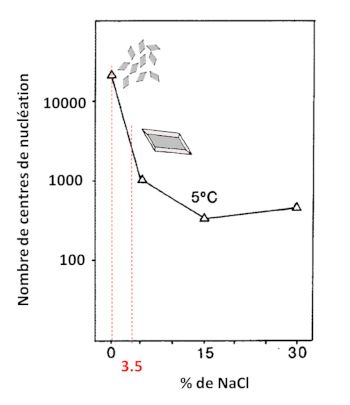
Source - © 2025 — Bernard Barailler d'après Cody et Cody, 1988 [ 7 ] , modifié
On observe, par exemple, que le passage d'une salinité nulle à celle de la mer (3,5 %) divise par 10 le nombre de centres de nucléation. Au maximum, la différence de salinité induit un rapport de 82. Ces centres de nucléation sont en compétition pour capter les unités de croissance présentes dans la solution. En première approche, la taille des cristaux est donc inversement proportionnelle à la densité de nucléation. Les résultats montrent que des cristaux de gypse beaucoup plus gros sont obtenus en présence de NaCl ajouté.
Dans les terrains présents sous le paléo-lac du Trièves, les évaporites du Trias et les marnes callo-oxfordiens peuvent être des sources d'apports d'eaux salées. Les variations de salinité du lac pourraient alors expliquer pourquoi, sur certaines strates, on a beaucoup de petits cristaux au mètre carré alors que d'autres strates comportent des cristaux plus gros avec une densité spatiale plus faible au mètre carré (la masse de gypse au mètre carré pouvant rester la même). Un raisonnement à la limite pourrait expliquer l'absence apparente de gypse dans certaines couches, car les cristaux seraient microscopiques. Ceci est une hypothèse qui reste à confirmer. On trouve à Sinard des cristaux de 10 cm et d'autres de 0,1 cm. Pour passer d'un cristal de 10 cm à son équivalent en cristaux de 0,1 cm, il faudrait 1 million (106) de petits cristaux pour garder la même quantité cristallisée. Donc la seule modification des apports de NaCl ne peut expliquer ces variations hypothétiques (rapport maximum de 82 sur le nombre de centre de nucléation), ni la présence de couches sans gypses visibles. Si la teneur en NaCl peut “aider” à diminuer le nombre de germes, une insuffisance en soluté pour former du gypse parait nécessaire, et comme le calcium est largement disponible, c'est la teneur en ions sulfate et/ou Mg2+ qui semble être le facteur “limitant”.
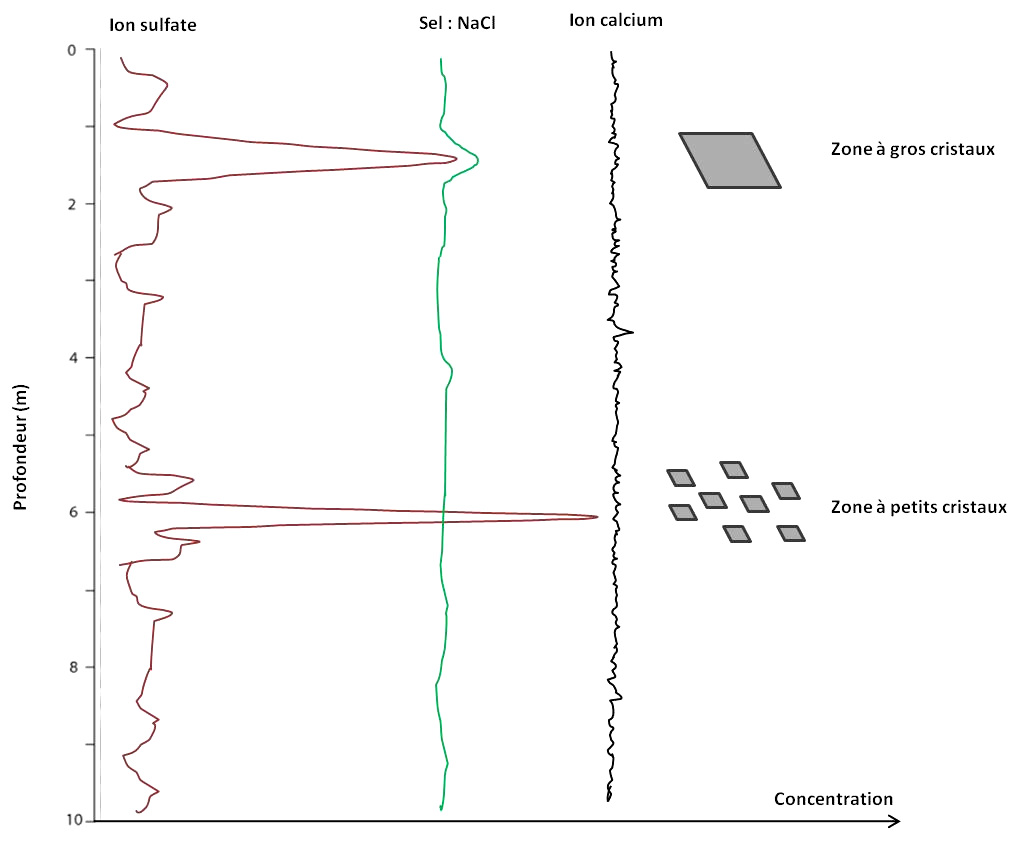
Des variations de concentration des ions au sein des varves
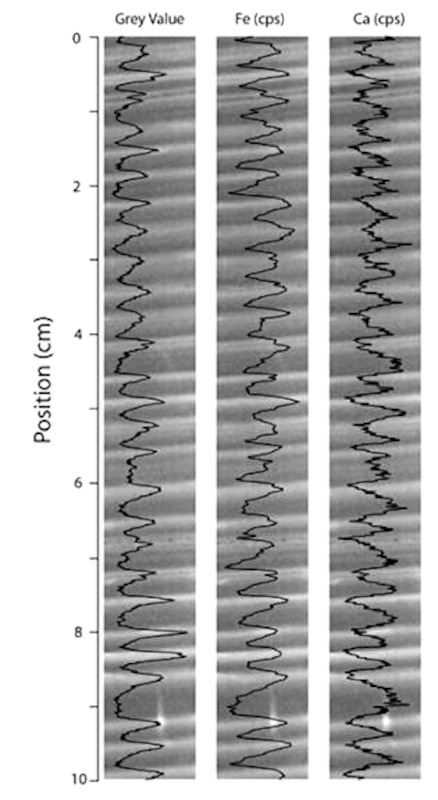
La teneur en ions dans les varves est variable. La figure suivante (figure 15) illustre un exemple des teneurs en calcium et fer le long d'une série de varves d'un lac canadien.
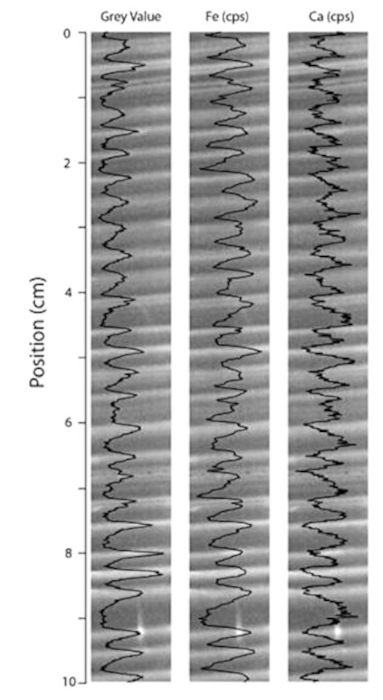
Source - © 2015 — D'après Zolitschka et al. [ 33 ] , modifié
On observe des couplets annuels clairs / sombres avec des niveaux clairs (estivaux) plus riches en fer que les niveaux sombres (printaniers) et l'inverse pour le calcium.
Dans le cas de la figure précédente, les varves sont caractérisées par des sous-lames printanières sombres et liées au ruissellement, qui sont riches en éléments allochtones notamment en Ca, tandis que les sous-lames pâles de l'été à l'automne sont enrichies en Fe. Les variations interannuelles sont très faibles (les mêmes variations saisonnières se répètent d'année en année). Les variations saisonnières étant bien conservées, on peut s'attendre à ce que les variations interannuelles soient elles aussi préservées et donc que de possibles enrichissements épisodiques en sodium et/ou en sulfate restent cantonnés dans les couches de varves correspondantes.
Dans cet esprit, avec une faible variation interannuelle de la teneur en calcium et une variation épisodique et plus ou moins importante des teneurs en sodium et sulfate, le schéma suivant (figure 16) modélise de manière simplifiée la présence et la taille des cristaux de gypse dans les varves.
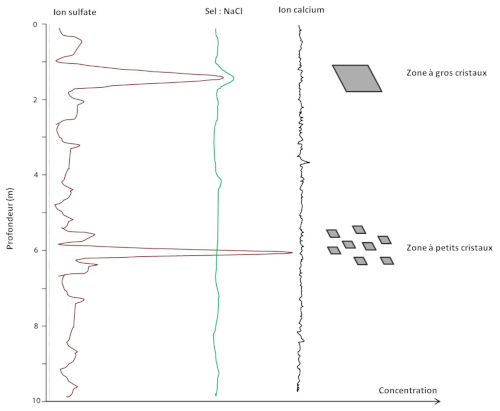
Source - © 2025 — Bernard Barailler
Les ions sulfate sont ici le facteur limitant. Leur absence ou leur faible teneur ne permet pas d'avoir de gypse, on a alors des couches stériles en gypse. Leur présence, en quantité “juste suffisante” ou en grande quantité, permet la sursaturation et la formation de gypses dont la taille pourrait dépendre d'autres facteurs tels que la teneur en ions sodium, dont la forte teneur favorise l'apparition d'un nombre réduit de cristaux potentiellement plus gros. Une faible teneur en sel augmente le nombre de germes et provoque la création de nombreux cristaux plus petits. Enfin, les ions Mg2+, non représentés sur la figure 16, sont aussi des inhibiteurs de genèse du gypse.
Conclusion, de la formation des gypses de Sinard à leur récolte
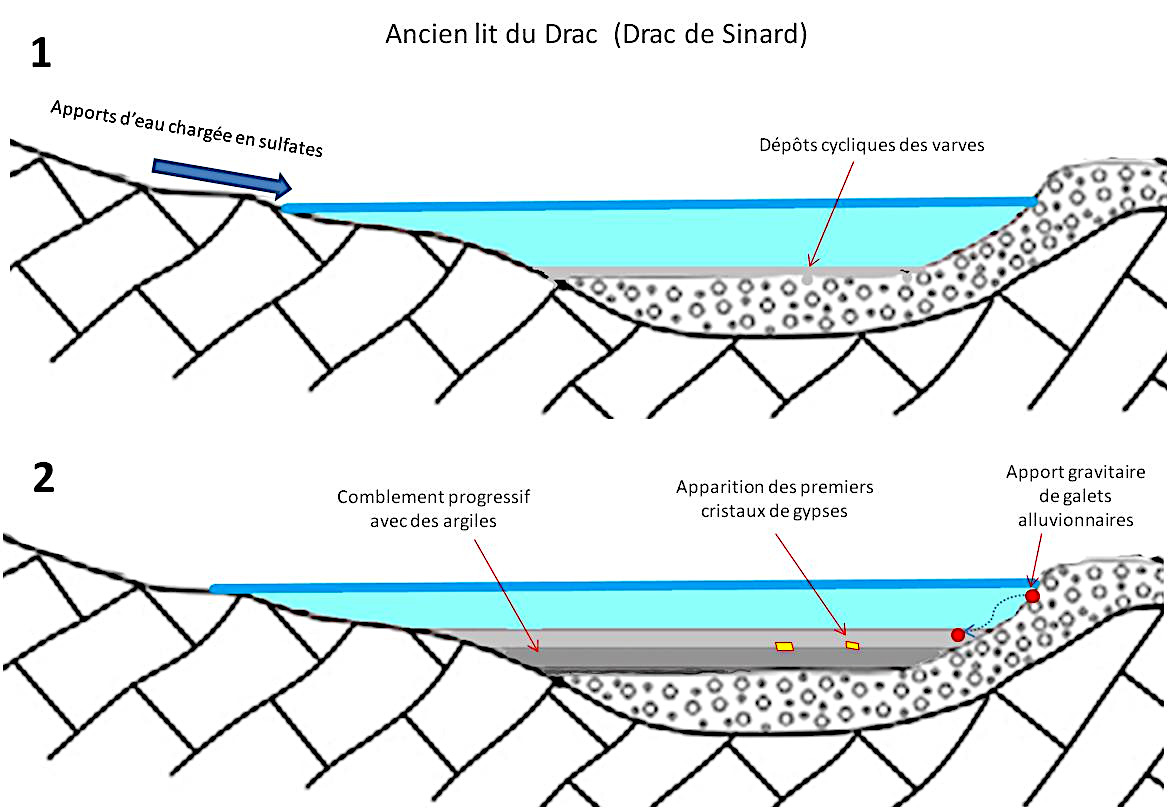
À partir de toutes les données rassemblées et des modèles proposés précédemment, voici une présentation simplifiée de la croissance des gypses de Sinard.
(1) À l'époque du Würm, un lac glaciaire se forme. Il est gelé l'hiver. Ceci amène un apport cyclique de sédiments qui vont former des varves. Par ailleurs les rivières apportent épisodiquement des ions sulfate issus de l'oxydation des sulfures des bassins versants et du lit des cours d'eau alimentant le lac (altération de l'Aalénien à nodules pyriteux).
(2) La sédimentation progressive, par pression et porosité, chasse l'eau des couches argileuses inférieures. Ceci augmente la concentration des ions dans la solution restante. Lorsqu'on franchit la courbe de solubilité (ou de nucléation spontanée), le gypse cristallise dans les argiles.
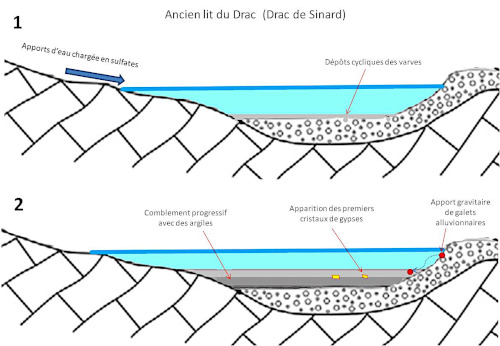
Source - © 2025 — Bernard Barailler
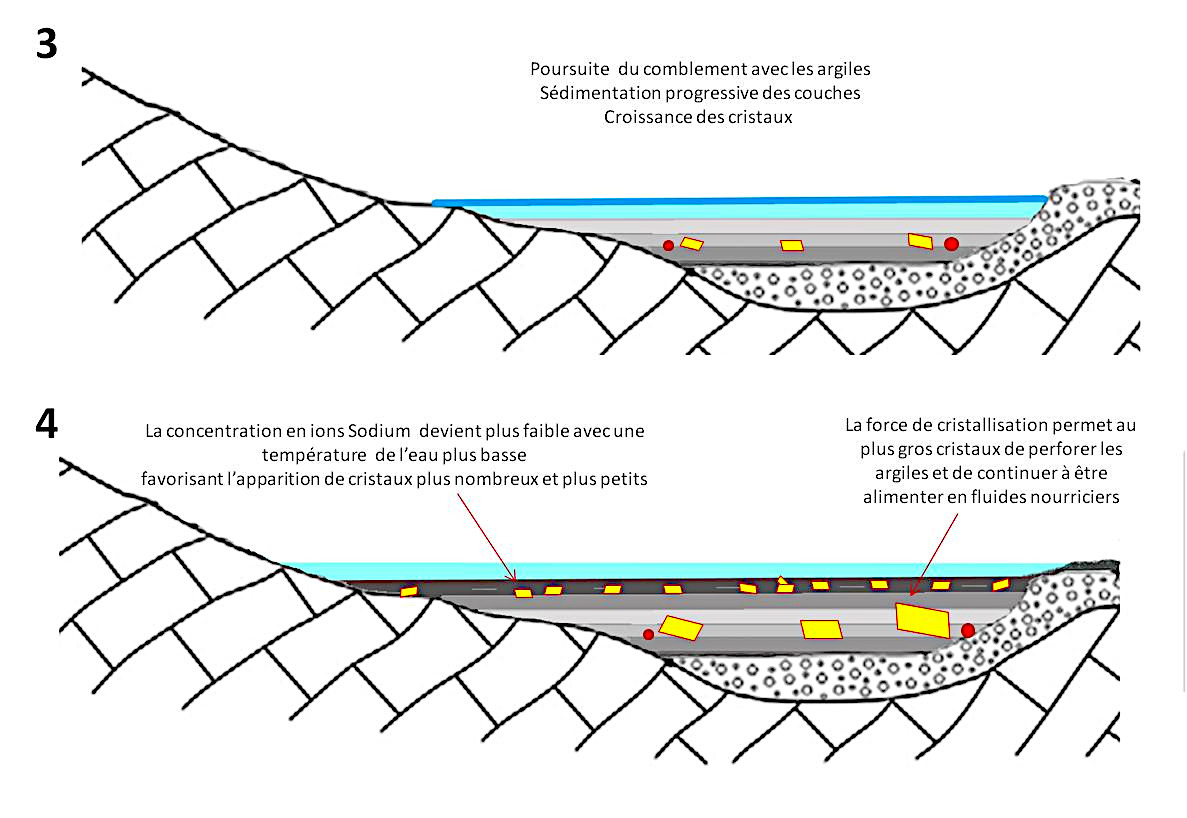
(3) La sédimentation se poursuit, des cristaux de gypse croissent en déformant les argiles par la force de cristallisation et atteignent leur taille définitive en quelques années. Parfois les conditions de cristallisation des gypses ne sont pas réunies (quantité insuffisante d'ions sulfate, présence d'ions inhibiteurs comme le magnésium…), les couches d'argile correspondantes ne présentent alors pas de cristaux visibles, du moins pas visibles à l'œil nu.
(4) Pendant la durée de présence du paléo-lac, la concentration en ions de l'eau a pu varier en fonction des couches géologiques traversées ou mises à l'affleurement. La concentration en ions sodium peut ainsi avoir varié. Des périodes plus froides ont pu favoriser la nucléation (légère baisse de solubilité) et ainsi l'apparition de cristaux plus nombreux (et plus petits). La présence de quelques galets peut ça et là générer des apports supplémentaires d'ions et favoriser la croissance de cristaux à proximité.
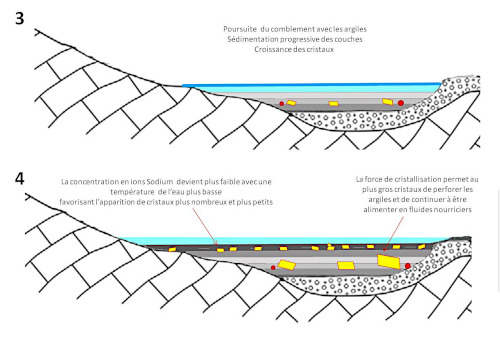
Source - © 2025 — Bernard Barailler
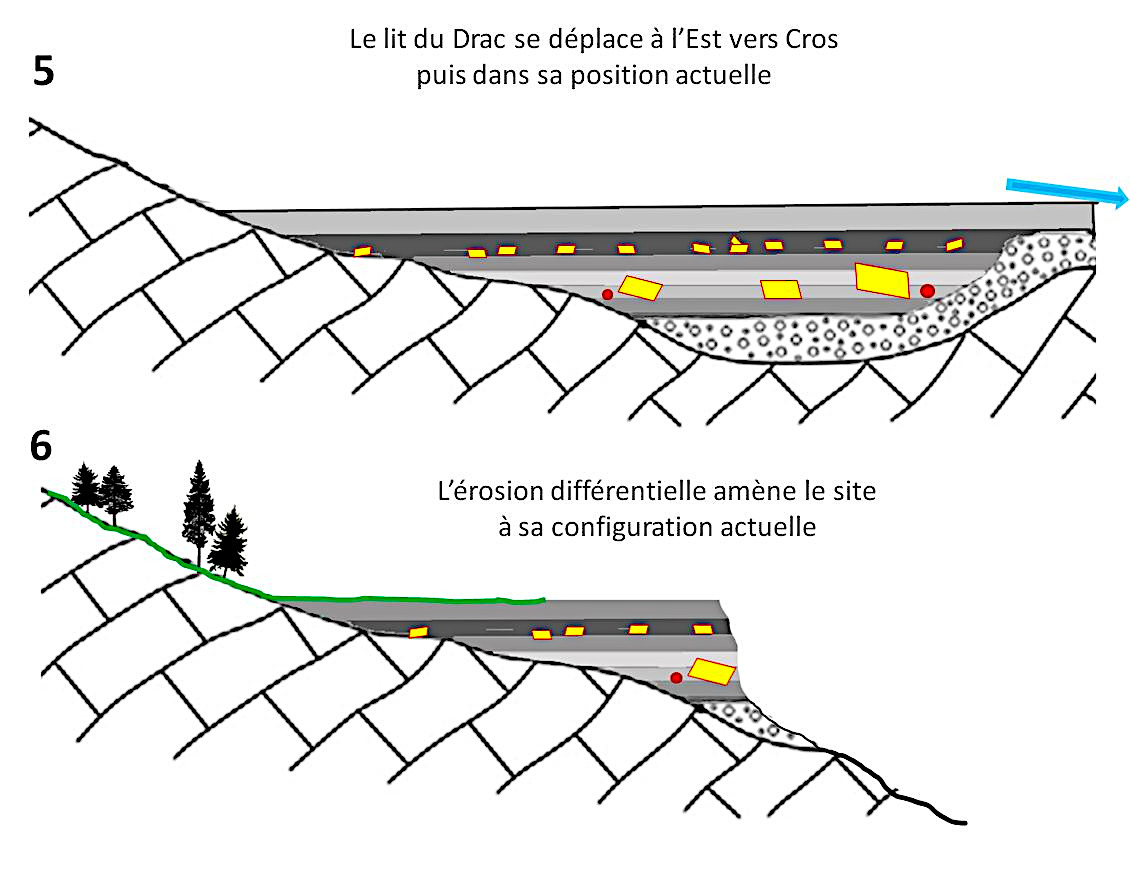
(5) Le lit du Drac évolue et se déplace vers l'Est. La zone s'assèche. la croissance des cristaux est à priori interrompue.
(6) L'érosion produit son œuvre, amenant le site à sa configuration actuelle. Sur le front de taille les couches de varves avec leurs cristaux apparaissent. Les cristaux qui sont à l'air libre sont progressivement dissouts. Ceux qui sont dans les argiles imperméables sont protégés. Ils exhibent alors parfois la forme d'un cristal monoclinique proche de la perfection (figure 1).
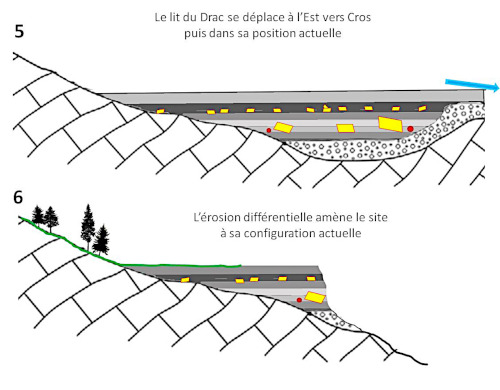
Source - © 2025 — Bernard Barailler
En plus des cristaux de gypse “exemplaires”, de magnifiques gypses à macles en forme de sapin (figure 20) peuvent également être trouvés dans ces argiles. Leur genèse sera présentée dans un article à venir.

Source - © 2025 — Bernard Barailler
Ce sapin de gypse mesure 8 cm de hauteur.
Remerciements
Le texte soumis a bénéficié des avis et commentaires d’Andrea Cotellucci (Labo. chimie et corrosion, Cogne Accial Speciali, Aoste, Italie), et d’une relecture de Jean-Jacques Aubert (LETI, CEA).
Olivier Dequincey (ENS de Lyon / DGESCO) est remercié pour sa relecture, sa contribution à la modélisation du produit de solubilité, et ses suggestions concernant les quantifications et arguments géologiques de terrain, pour aboutir au texte publié.
Bibliographie
- E.A. Abdel-Aal, H.M. Abdel-Ghafar, B.E. El Anadouli, 2015. New Findings about Nucleation and Crystal Growth of Reverse Osmosis Desalination Scales with and without Inhibitor(lien externe - nouvelle fenêtre), Crystal Growth & Design, 15, 10, 5133-5137
- S. Ben Ahmed, M.M. Tlili, M. Amami, M. Ben Amor, 2014. Gypsum precipitation kinetics and solubility in the NaCl-MgCl2-CaSO4-H2O system(lien externe - nouvelle fenêtre), Industrial & Engineering Chemistry Research, 53, 23, 9554-9560 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- B. Barailler, 2024. Comment se forment les cristaux ? Du bécher à la croissance hydrothermale, Planet Terre - ISSN 2552-9250
- B. Barailler, 2025. La force de cristallisation, ou quand les gypses déchirent les varves du Trièves !, Planet Terre - ISSN 2552-9250
- S.C. Chapra, A., G.J. Warren, 2012. Long-term trends of Great Lakes major ion chemistry(lien externe - nouvelle fenêtre), Journal of Great Lakes Research, 38, 3, 550-560 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- Q. Chen, L. You, Y. Kang, L. Dou, J.J. Sheng, 2018. Gypsum-Crystallization-Induced Fracturing during Shale−Fluid Reactions and Application for Shale Stimulation(lien externe - nouvelle fenêtre), Energy & Fuels, 32, 10, 10367-10381
- R.D. Cody, A.M. Cody, 1988. Gypsum nucleation and crystal morphology in analog saline terrestrial environments(lien externe - nouvelle fenêtre), Journal of Sedimentary Research, 58, 2, 247-255
- O. Dequincey, 2025. Solubilité et solutions aqueuses, rappels et calculs, Planet Terre - ISSN 2552-9250
- J. Gaidis, 2020. How does electrolytic conduction depends on the size of ions & their solvation?(lien externe - nouvelle fenêtre), Chemistry-StackExchange (consulté le 15 mai 2025)
- J. Garside, A. Mersmann, J. Nyvlt (eds), 2003. Measurement of crystal growth and nucleation rates - 2nd edition, Institution of Chemical Engineers, 202p
- Geilfus N.-X., R.J. Galley, M. Cooper, N. Halden, A. Hare, F. Wang, D.H. Søgaard, S. Rysgaard, 2013. Gypsum crystals observed in experimental and natural sea ice(lien externe - nouvelle fenêtre), Geophysical Research Letters, 40, 6362-6367 [Free Access]
- A. Giraud, P. Antoine, T.W.J. Van Asch, J.D. Nieuwenhuis, 1991. Geotechnical problems caused by glaciolacustrine clays in the French Alps(lien externe - nouvelle fenêtre), Engineering Geology, 31, 2, 185-195
- X. Guillot, M. Al-Mukhtar, F. Bergaya, J.-M. Fleureau, 2002. Estimation de la porosité dans un matériau argileux(lien externe - nouvelle fenêtre), Comptes Rendus Geoscience, 334, 2, 105-109 [Open archive]
- D. Jongmans, G. Bièvre, F. Renalier, S. Schwartz, N. Beaurez, Y. Orengo,, 2009. Geophysical investigation of a large landslide in glaciolacustrine clays in the Trièves area (French Alps)(lien externe - nouvelle fenêtre), Engineering Geology, 109, 1-2, 45-56 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- F. Lapointe, 2012. Reconstitutions paléoclimatiques à partir de la technique d'analyse d'images sur les sédiments varvés de Cape Bounty, île Melville, Canada(lien externe - nouvelle fenêtre), Mémoire de Maîtrise en sciences de la terre, Université du Québec - INRS, 134 p [Accès libre]
- A.S. Larsen, J.A. O'Donnell, J.H. Schmidt, H.J. Kristenson, D.K. Swanson, 2017. Physical and chemical characteristics of lakes across heterogeneous landscapes in arctic and subarctic Alaska(lien externe - nouvelle fenêtre), Journal of Geophysical Research : Biogeosciences, 122, 4, 993-1008 [Open Access]
- M.G. Loso, 2008. Summer temperatures during the Medieval Warm Period and Little Ice Age inferred from varved proglacial lake sediments in southern Alaska(lien externe - nouvelle fenêtre), Journal of Paleolimnology, 41, 1, 117-128 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- D.B. Maier, J. Rydberg, C. Bigler, I. Renberg, 2013. Compaction of recent varved lake sediments(lien externe - nouvelle fenêtre), GFF, 135, 3-4, 231-236 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- Y. Marcus, 1998. The Properties of Solvents(lien externe - nouvelle fenêtre), Wiley, 256p [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- G.D. Matheson, G.L. Jones, 2015. The habit and form of gypsum crystals in Irish mudstone aggregate affected by pyrite-induced swelling(lien externe - nouvelle fenêtre), Quarterly Journal of Engineering Geology and Hydrogeology, 48, 3-4, 167-174
- M. Oshchepkov, K. Popov, A. Kovalenko, A. Redchuk, J. Dikareva, I. Pochitalkina, 2020. Initial Stages of Gypsum Nucleation: The Role of “Nano/Microdust”(lien externe - nouvelle fenêtre), Minerals, 10, 12, 1083 [Open Access]
- M. Pelser, J.J. Eksteen, L. Lorenzen, C. Aldrich, 2003. The control of calcium and magnesium in base metal sulphate solutions(lien externe - nouvelle fenêtre), Proceedings: XXII International Mineral Processing Congress, 1240-1248 [pdf]
- C. Pisapia, 2006. Identification de bio-signatures structurales, chimiques et isotopiques à l'interface bactérie/minéral dans le système Acidithiobacillus ferrooxidans/pyrite(lien externe - nouvelle fenêtre), thèse de doctorat, Institut National Polytechnique de Lorraine, 364p
- J.-L. Probst, A. Probst, 2015. Composition chimique des eaux et variabilité naturelle(lien externe - nouvelle fenêtre), in A. Euzen, C. Jeandel, R. Mosseri (éd.), L'eau à découvert(lien externe - nouvelle fenêtre), CNRS Éditions, 206-207 [Accès ouvert freemium]
- F. Rahman, 2013. Calcium sulfate precipitation studies with scale inhibitors for reverse osmosis desalination(lien externe - nouvelle fenêtre), Desalination, 319, 79-84 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- E. Roedder, 1969. Varvelike banding of possible annual origin in celestite crystals from Clay Center, Ohio, and in other minerals(lien externe - nouvelle fenêtre), American Mineralogist, 54, 5-6, 796-810 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- Y. Rong, 2018. Formation du sulfate de calcium hémihydrate de type α à partir de gypse par un procédé de dissolution-cristallisation : étude cinétique expérimentale et modélisation(lien externe - nouvelle fenêtre), thèse de doctorat, Univ. Paris sciences et lettres, 244p
- M.-A. Selosse, D. Busti, P. Thomas, 2008. Les sources thermominérales d'Auvergne : chimiolithotrophie et photosynthèse, Planet Terre - ISSN 2552-9250
- A.A. Shaikh, I.W. Kazi, F. Rahman, 2015. Synthesis and evaluation of phosphate-free antiscalants to control CaSO4·2H2O scale formation in reverse osmosis desalination plants(lien externe - nouvelle fenêtre), Desalination, 357, 36-44 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
- P. Thomas, 2021. Les gypses automorphes des lacs glaciaires würmiens du Trièves, Cornillon-en-Trièves, Sud de l'Isère, Planet Terre - ISSN 2552-9250
- M.J. Waker, R.D. Robarts, 1995. Microbial nutrient limitation in prairie saline lakes with high sulfate concentration(lien externe - nouvelle fenêtre), Limnology and Oceanography, 40, 3, 566-574 [Free Access]
- H. Yuan, J. Liu, Q. Ding, Z. Liu, Y. Wen, 2025. A thermodynamic general prediction model for the solubility of sulfate scale minerals in pure water at high temperature and high pressure(lien externe - nouvelle fenêtre), Journal of Molecular Liquids, 422, 126930
- B. Zolitschka, P. Francus, A.E.K. Ojala, A. Schimmelmann, 2015. Varves in lake sediments – a review(lien externe - nouvelle fenêtre), Quaternary Science Reviews, 117, 1-41 [PDF – Texte intégral(lien externe - nouvelle fenêtre)]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}